 Koordinationschemie II (AC7–9) Koordinationschemie II (AC7–9)
Koordinationschemie II (AC7–9) Koordinationschemie II (AC7–9)2 SWS,
Fr 10–12, Willstätter-Hörsaal,
Beginn: 23. Oktober 2009
Die Klausur für das WS 2009/2010 findet am Freitag, dem 12. Februar 2010, um 10:15 Uhr statt; Zeit: 1 h, Ort: Willstätter-Hörsaal. Bitte melden Sie sich an.
Alte Klausuren:
| WS 2004/2005: | Klausur | Lösung | Ergebnis |
| WS 2005/2006: | Klausur | Lösung | Ergebnis |
Master-Studiengang Chemie: AC 7, 8 oder 9
Der Vorlesung Koordinationschemie II vertieft den Stoff der Vorlesung Einführung in die Koordinationschemie in verschiedene Richtungen. Ziel ist es, unter besonderer Berücksichtigung der verwendeten Methoden aktuelle Schwerpunkte der Koordinationschemie anzusprechen.
Prüfen Sie hier, ob Ihr Browser das Skript korrekt darstellt.
Zuletzt geändert: Januar 2010.
Die erste Lehreinheit knüpft an den Inhalt der Lehreinheit 2 der Einführung in die Koordinationschemie an. Es geht um die Frage, wie ein Komplexgleichgewicht in wässriger Lösung analysiert werden kann. Im Mittelpunkt steht als wohlvertrauter Ligand Tartrat, und zwar das C2-symmetrische l-Tartrat, das in mehr oder weniger alkalischer Lösung eingesetzt wird. Um übersichtlich zu bleiben, wird ein Palladium(II)-Zentrum mit eingeschränkter Funktionalität eingesetzt: zwei der vier (warum vier?) Bindungsstellen des Zentralmetalls sind durch einen zweizähnigen Stickstoff-Chelatliganden blockiert. Bei der Umsetzung des Edukts [(R,R-chxn)PdII(OH)2] (chxn = 1,2-Diaminocyclohexan) mit Weinsäure (H2tart) in wässrig-alkalischer Lösung entstehen neue Komplexverbindungen. Da diese diamagnetisch sind (warum?), können 13C-NMR-Spektren mit Standardmethoden ausgewertet werden (1H-NMR-Spektren helfen nicht viel weiter; warum nicht?).
Auch als pdf verfügbar (rechts anklicken und entscheiden, was man will): 13C-NMR Spektren in D2O bei verschiedenen molaren Verhältnissen [(R,R-chxn)Pd(OH)2]:l-H2tart:OH− von 1:1:1, 3:1:0, 1:1:2 und 3:1:2. Die auf der Abszisse angegebene chemische Verschiebung bezieht sich auf TMS (δ = 0 ppm). Spektrum a: Dinatrium-l-tartrat in D2O; Komplexspezies sind farbig dargestellt.
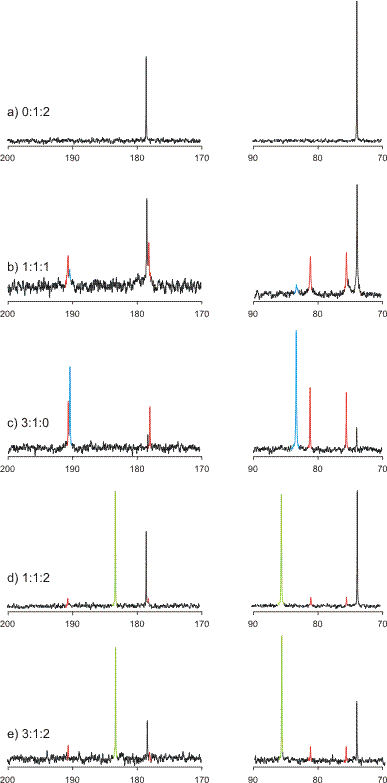
Die Spektren werden auf folgende Aspekte hin untersucht:
• Welche Komplexspezies entstehen?
• Wodurch wird gesteuert, welche Spezies dominiert?
• Wie kann die Metallbindungsstelle identifiziert werden? Stichwort: Coordination induced shift (CIS)
Die wichtigste und zur nächsten Lehreinheit überleitende Frage ergibt sich jedoch aus der Betrachtung der Spektren d und e: Wie ist es zu verstehen, dass ein Gleichgewicht wie
L2Pd2(tartH−2) + 2 OH− = LPd(tartH−2) + LPd(OH)2
nach dem 13C-NMR-Spektrum praktisch vollständig auf der rechten Seite liegt, dass also der hoch eingeschätzte Chelateffekt Schwächen zeigt (L = R,R-chxn)? Die oft als chaotisch empfundene Chemie in wässriger Lösung zeigt hier eine wesentliche Besonderheit: die erwarteten Komplexgleichgewichte spielen mit Protolysegleichgewichten zusammen. Liegt, wie im Beispiel, auf der einen Seite des Gleichgewichts starke Säure oder Base frei vor, so gewinnt die Natur bei der Verschiebung des Gleichgewichtes auf die andere Seite Neutralisationsenthalpie. Dieser Beitrag kann entscheidend sein, wenn die Komplexstabilität nicht sehr hoch ist – so wie hier: beachten Sie die großen Mengen an freiem Ligand zum Beispiel in Spektrum b.
Der Aspekt des Lösungsgleichgewichts bekommt in dieser Lehreinheit eine noch größere Bedeutung. Wir stecken uns das Ziel, einen Mehrkernkomplex aufzubauen, und schaffen damit die Grundlage zum Aufbau interessanter Materialien, zum Beispiel molekularer Magnete. In dieser Lehreinheit geht es sehr „un-organisch“ zu. Sowohl in der Organischen Chemie als auch in der Anorganischen Molekülchemie bauen Sie Moleküle schön peu-a-peu in Einzelschritten auf, indem Sie ausnutzen, dass Ihr jeweiliges Zwischenprodukt durch hinreichend hohe Aktivierungsbarrieren darin gehindert wird, irgendetwas zu tun, was Sie nicht wollen – Sie bauen an kinetisch inerten Molekülen herum. Dieser Weg ist in der Koordinationschemie oft verschlossen. Viele wichtige Metallzentren sind bezüglich des Ligandaustauschs kinetisch labil. Deren Chemie verläuft unter strikter thermodynamischer Kontrolle, meist ohne dass Aktivierungsbarrieren erkennbar sind. Wir lernen eine wichtige Methode kennen, unter dieser Randbedingung komplexe Strukturen aufzubauen: die Kontrolle durch die Stöchiometrie und den pH-Wert der Reaktionslösungen.
Konkret schauen wir folgendes Beispiel an: Wir vereinfachen Tartrat zu Tartronat (das ist das Anion der 2-Hydroxy-1,3-propandisäure, also Hydroxymalonat, wenn Sie so wollen) und setzen mindestens die doppeltmolare Menge dieses „ambidenten“ Liganden in wässrig-neutraler Lösung mit Kupfer(II)-Salzen um. Es lassen sich dann Salze wie zum Beispiel Li2[Cu(C3H2O5)2(H2O)2] · 2 H2O isolieren. Die Struktur der Diaqua-bis(tartronato-O1,O3)-cuprat(II)-Dianionen ist nicht ungewöhnlich: über die Carboxylatfunktionen der Tartronat-Liganden sind sechsgliedrige Chelatringe gebildet worden. Die Cu-O-Abstände betragen ca. 195 pm (1.95 Å). Mit 291 pm deutlich weiter entfernt sind zwei O-Atome von Aqualiganden. Das Tartronatocuprat zeigt damit die zu erwartende Jahn-Teller-Verzerrung in ungewöhnlich deutlicher Form. Die Ursache hierfür wird weiter unten behandelt.
Als nächstes wird nun gespart, und zwar am Liganden. Es wird nur soviel Tartronat eingesetzt, dass sich ein Ligand:Metall-Verhältnis von 1:1 ergibt. Der pH-Wert muss nun mit etwas Base auf 7 eingestellt werden, um wieder eine klare Lösung zu erhalten. Wir schauen uns wieder die Struktur eines aus solchen Lösungen isolierten kristallinen Produkts an, zum Beipsiel Cs3[Cu3(C3HO5)3(H2O)3] · H2O: es ist ein dreikerniger Komplex entstanden. Die Regel, dass ein verrringertes Ligandangebot zum Brückenbindungsmodus, also zur Verteilung der nun knapperen Lewis-Basizität kommt, ist ein allgemeines Prinzip. Man denke nur an die Reaktionen von zum Beispiel AlCl3 in aprotischen Lösungsmitteln: Äquimolare Mengen AlCl3 und Cl− führen zu [AlCl4]−; wird nur die halbe molare Chloridmenge zur Verfügung gestellt, entsteht das Anion [Al2Cl7]− mit einem verbrückenden Chloridoliganden gemäß [Cl3Al–Cl–AlCl3]−. Wir fragen uns, welche Regel hinter dieser Beobachtung steckt; außerdem fragen wir uns (nochmal), warum eine pH-Wert-Erhöhung bei Tartronat-Überschuss zu denselben Komplexen führt. Das heißt, wir schauen das Gleichgewicht an:
3 [Cu(C3H2O5)2]2− + 3 OH− = [Cu3(C3HO5)3]3− + 3 C3H2O52−
Nachdem die thermodynamischen Randbedingungen geklärt sind, lautet die nächste Frage, ob besondere Eigenschaften für das dreikernige Cuprat(II) zu erwarten sind. Die Strukturdaten zeigen günstige Voraussetzungen für Superausstausch an. Die bei Kupfer(II)-Komplexen sehr häufige antiferromagnetische Kopplung sollte bei dem betrachteten Tricuprat zu „Spinfrustration“ führen. die Winkel an O zeigen allerdings (wie fast immer) eine Aufhebung der C3-Symmetrie an (Cu-O-Cu-Winkel vom rechten oberen Brücken-O-Atom im Uhrzeigersinn: 130, 129, 115°). Die Spinkopplung kann über längere Strecken vermittelt werden. Ein Lehrbuchbeispiel ist Kupfer(II)-acetat, bei dem sich die Frage stellt, ob nicht eine Kupfer-Kupfer-Bindung die bessere Erklärung für den beobachteten S=0-Grundzustand ist.
Auch als pdf: Ausschnitt aus den Kristallstrukturen von Li2[Cu(C3H2O5)2(H2O)2] · 2 H2O und Cs3[Cu3(C3HO5)3(H2O)3] · H2O:
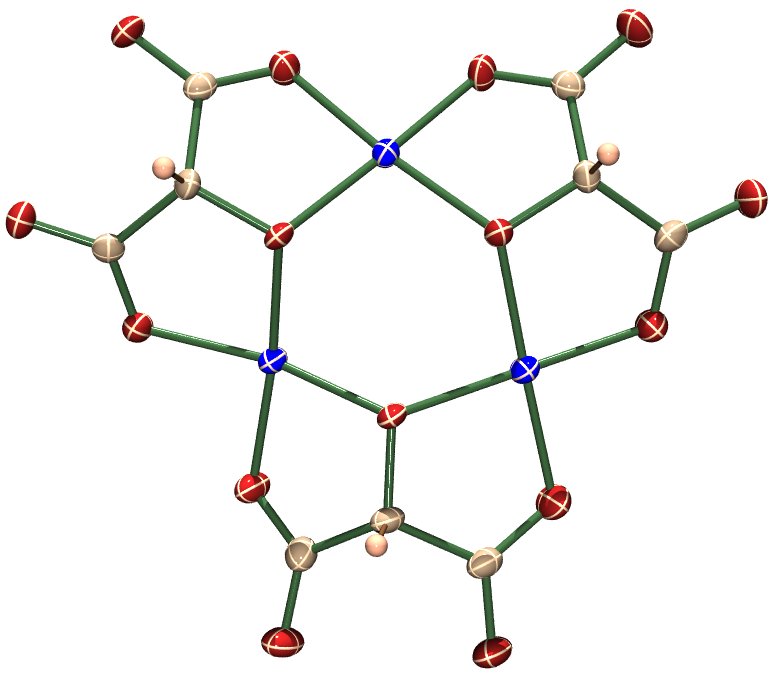
Die Spins der paramagnetischen Zentralmetallatome wechselwirken in Mehrkernkomplexen über chemische Bindungen hinweg oder direkt aufgrund ihrer räumlichen Nähe. Die häufigste Wechselwirkung ist die antiferromagnetische Kopplung, bei der sich die Spins benachbarter Zentren im Grundzustand antiparallel ausrichten. Der übliche Kopplungsweg ist der Superaustausch entlang eines Pfades CuA–O–CuB, wenn mit Kupfer und μ-Oxido-Liganden formuliert wird. Das spintragende Orbital des Kupfers ist im Tartronato-tricuprat das x2 − y2-Orbital (warum?). Wird CuA α-Spin zugeordnet, so wird β-Spin in den unmittelbar benachbarten Lappen eines p-Orbitals des μ-O-Atoms induziert und damit α-Spin in den abgewandten Orbitallappen. Dieser α-Spin induziert dann wieder β-Spin in das magnetische Orbital von CuB – die beiden Metallzentren sind antiferromagnetisch gekoppelt. Das Ausmaß der Kopplung wird üblicherweise durch die Kopplungskonstante J ausgedrückt, die die Energie für die Entkopplung der Spins an den Zentren A und B beschreibt. Ein negativer Wert für J entspricht einem S=0-Grundzustand, also der antiferromagnetischen Kopplung (cave: diese Regelung wird in der Literatur nicht einheitlich angewendet!). In der Chemie wird J üblicherweise in cm−1 angegeben. Bei dem beschriebenen Superaustauschpfad ist der Betrag von J umso größer, je stumpfer der Cu-O-Cu-Winkel ist. Solche Beziehungen werden durch die Goodenough-Kanamori-Regeln beschrieben, auf die weiter unten ausführlicher eingegangen wird. Werden zwei Kupfer(II)-Zentren durch Sauerstoff-Liganden verbrückt, sollte bei Cu-O-Cu-Winkeln um 110–115° antiferromagnetische Kopplung einsetzen. Bei Werten um 140° wird eine so große Kopplungkonstante erwartet, dass der angeregte Triplettzustand auch bei Raumtemperatur kaum populiert ist.
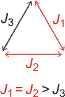
Im vorliegenden Beispiel ist ein besonders interessanter Fall nicht realisiert. Wäre das Tricuprat-Ion C3-symmetrisch, so wäre J1 = J2 = J3. Durchgehende antiferromagnetische Kopplung wäre dann nicht möglich, es läge „Spinfrustration“ vor.
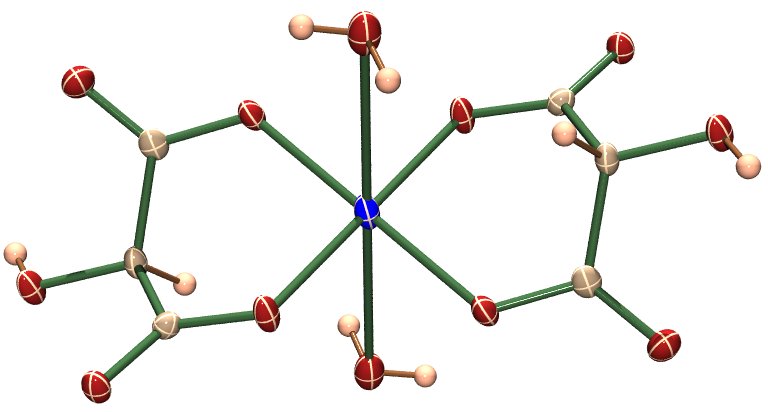
Die zuletzt besprochenen Komplexe waren zugleich (kinetisch) labil und (thermodynamisch) stabil. Mit Komplexen, die diese Randbedingung erfüllen, wurde die heute besonders aktuelle Teildisziplin der Supramolekularen Chemie begründet (Jean-Marie Lehn, Nobelpreis 1987). Das Grundprinzip wird besonders prägnant bei Lehns „Helicaten“ deutlich. Deren Baustein ist zum Beispiel das Komplexkation Bis(2,2'-bipyridyl)-kupfer(I), [Cu(bpy)2]+, das den zweizähnigen bpy-Chelatliganden enthält. [Cu(bpy)2]+ kann mit einem geeigneten Gegenion in methanolischer Lösung hergestellt werden. (Was darf erwartet werden, wenn Wasser als Reaktionsmedium gewählt wird?) Als Komplex eines d10-Zentralmetalls mit vier Ligatoratomen ist tetraedrischer Aufbau zu erwarten (warum nicht quadratisch-planarer?). Mit diesem Bauelement werden nun mehrkernige Komplexe (mit viel größeren Metall-Metall-Abständen als bei den Tartronato-cupraten) aufgebaut, indem bpy-Moleküle durch eine flexible Brücke zusammengefügt werden. Das einfachste Beispiel ist:
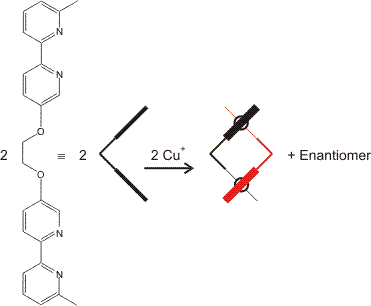
Drei oder vier Metallzentren lassen sich in analoger Weise in eine Helix einbauen. Ein besonders lehrreicher Fall liegt vor, wenn verschiedene Helicat-Bildner gemischt und dann mit der benötigten Mengen Kupfer(I) umgesetzt werden. Das folgende Schema zeigt das Ergebnis:
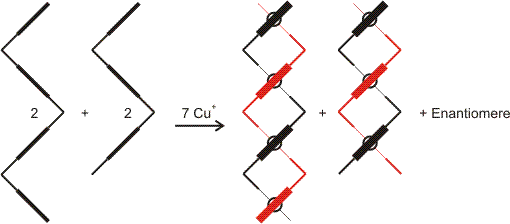
Es wäre zu erwarten, dass ein Gemisch mit zahlreichen Komplexen entsteht, was aber nicht der Fall ist. Die Ursache ist die kinetische Labilität der Kupferzentren bezüglich des Ligandaustauschs. Natürlich entstehen bei der Umsetzung von Ligandgemisch und Kufper(I)-Salz auch Komplexe, die unerwünschten Kombinationen entsprechen. Ein Beispiel könnte so aussehen:

Es wird deutlich, dass durch die Fehlpassung nicht alle Metallbindungsstellen des Liganden besetzt sind. Da in ortho-Stellung zum ersten und letzten N-Atom eines jeden Liganden eine Methylgruppe verhindert, dass Polymerisation eintritt, ist dieser Ausweg verschlossen, die noch fehlende Bindungsenergie zu nutzen. Unter den gegebenen Versuchsbedingungen ist jedoch eine Reparatur der Fehlpassung möglich. Das kinetisch labile System nähert sich so allmählich der Belegung aller Metallbindungsstellen, also dem ausgeordneten, thermodynamisch stabilen Zustand. (Die letzten 3 Bilder als pdf.)
Dasselbe Grundprinzip, das zu Helicaten führt, eröffnet auch einen besonders aktuellen Zweig der Supramoleklaren Chemie, bei dem Metallatom-Gitter aufgebaut werden. Anstelle der eindimensionalen Ordnung des Helicats wird hier eine zweidimensionale Ordnung aufgebaut [supra1].
M. Ruben, J. Rojo, F. J. Romero-Salguero, L. H. Uppadine, J.-M.. Lehn: Metallionen-Gitterarchitekturen: funktionelle supramolekulare Metallkomplexe. Angew. Chem. 2004, 116, 3728–3747 [supra1].
Paramagnetische Metallzentren sind bevorzugte Motive, die in supramolekulare Strukturen einbaut werden. Dahinter steht die Hoffnung, magnetische Wechselwirkungen in ausgedehnten, aber wohldefinierten Strukturen zu erzeugen. In dieser Lehreinheit wird eine grundlegende Frage der Koordinationschemie beleuchtet: Führt ein räumlich enger Kontakt paramagnetischer Zentren zu einer magnetischen Wechselwirkung? – zu einer Bindung?
Als Vorübung sehen wir uns die elektronische Struktur eines paramagnetischen Komplexes an, des [Mn(H2O)6]2+-Ions.
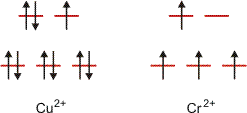
Um nebenbei koordinationschemische Grundregeln aufzugreifen, werden strukturell und chemisch nah verwandte Verbindungen betrachtet: Kupfer(II)- und Chrom(II)-acetat. Generell gilt, dass Kupfer(II)- und Chrom(II)-Verbindungen strukturell eng verwandt sind. Viele Salze sind isotyp. Ursache der Ähnlichkeit ist eine gemeinsame Besonderheit der d9- bzw. der high-spin-d4-Konfiguration. Beides sind bei oktaedrischer Koordination Jahn-Teller-Ionen, deren energetisch entartete Konfigurationen sich in der Besetzung des eg-Niveaus unterscheiden. Die Aufhebung der Degeneration bei diesen direkt auf die Liganden gerichteten Orbitale geht mit einer deutlichen Verzerrung des Koordinationsoktaeders einher, meist zu einer gestreckten quadratischen Bipyramide (Oh → D4h).
Ein Paar besonders interessanter Strukturen stellen die beiden Acetate von Kupfer(II) und Chrom(II) dar. Chrom(II)-acetat entsteht bei der Umsetzung von zum Beispiel Chrom(II)-sulfat, das bei der Umsetzung von hochreinem(?) Chrom mit hochreiner(?) Schwefelsäure entsteht, mit Natriumacetat. Aus dem blassblauen Hexaqua-chrom(II)-Ion entsteht rotes Chrom(II)-acetat-Monohydrat. Bei der Strukturanalyse überrascht der recht kurze Cr-Cr-Abstand von 2.36 Å.
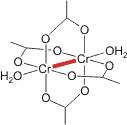
Werden die Aqua-Liganden entfernt, sinkt der ohnehin schon kurze Abstand drastisch. Für die Gasphase ergibt die Elektronenbeugung an [Cr2(AcO)4] 1.96 Å. In kristallinem wasserfreien Chrom(II)-acetat beträgt der Cr-Cr-Abstand 2.29 Å (können Sie die merkliche Differenz zwischen Gasphase und Kristall erklären?). Wird Acetat durch andere verbrückende Liganden ersetzt, so lässt sich der Metall-Metall-Abstand auf bis zu 1.828 Å verkürzen – dem kürzesten heute bekannten Abstand zweier Metallatome überhaupt. Die Vorstellung einer kovalenten Bindung zwischen den Chrom-Atomen wird vor allem durch Verbindungen gestützt, bei denen eine Cr2-Einheit nicht durch Liganden überbrückt ist. So wird in einem ebenfalls roten und diamagnetischen Dichromat [Cr2R6]2− mit R = N,N-Dimethylaminomethyl ein Abstand von 1.84 Å zwischen den Chromatomen gefunden (Organometallics 1998, 17, 475–478). Wie sollte eine solche Bindung aussehen? Werden zwei isolierte, quadratisch-planare CrO4-Fragmente betrachtet, die in z-Richtung keine weitere Liganden tragen, so befinden sich die 4 Valenzelektronen in den d-Orbitalen xy, xz, yz und z2. Das bei quadratisch-planarer Koordination instabilste x2−y2-Orbital bleibt unbesetzt. Nähern sich die beiden Cr-Atome entlang z an, so kommt es zuerst zur Überlappung der z2-Orbitale, also zu einer σ-Bindung. Diese wird durch weitere Bindungen unterstützt, für deren Bildung symmetrie-geeignete Orbitale zur Verfügung stehen: zwei entartete π-Bindungen (xz ↔ xz und yz ↔ yz) sowie eine δ-Bindung aufgrund der Wechselwirkung der beiden xy-Orbitale. Die rote Farbe der Dichrom(II)-Verbindungen beruht auf einem δ→δ*-Übergang. Eine Bindungsordnung von 4 ist bei den Chrom-Verbindungen im großen und ganzen akzeptiert, auch wenn es Gegenargumente gibt. Man beachte, dass der gemessene Diamagnetismus auch erklärt werden kann, wenn zum Beispiel allein von einer σ-Bindung ausgegangen wird und die übrigen Spins durch Austausch-Kopplung zum S=0-Grundzustand führen.
Bei der ersten Metall-Metall-Vierfachbindung, die in der Literatur beschrieben ist, bestehen diese Zweifel nicht. Im Octachlorido-dirhenat(III), [Re2Cl8]2−, in dem die Rhenium-Atome trotz ihrer Stellung in der dritten Übergangsreihe nur 2.24 Å voneinander entfernt sind (vgl. 2.75 Å in Rhenium-Metall), liegen die Chlorido-Liganden in der sterisch ungünstigen ekliptischen Konformation vor, in der ihr Abstand kleiner als die Summe der van-der-Waals-Radien ist. Nur in dieser Anordnung ist eine δ-Bindung möglich. Wird in die langwellige Absorption, dem δ→δ*-Übergang, eingestrahlt, so kommt es im angeregten Zustand zur Rotation in die gestaffelte Konformation.
Gibt es eine Analogie zwischen Chrom(II)-acetat-Monohydrat und einem entsprechenden Kupfer(II)-acetat-Monohydrat? Auf den ersten Blick unbedingt. Die Strukturen weisen die gleiche Konnektivität und Symmetrie auf. Lediglich der Metall-Metall-Abstand ist bei der Kupferverbindung mit 2.64 Å weniger spektakulär (vgl. 2.56 Å im Metall) – was aber auch nicht zu erwarten ist, da bestenfalls eine Einfachbindung gebildet werden kann.
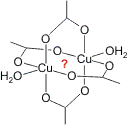
Eine Betrachtung des Kristallfeldschemas für ein quadratisch-planares CuO4-Fragment zeigt, dass das spintragende Orbital das x2−y2-Orbital ist. Die in Frage kommende Einfachbindung wäre also eine δ-Bindung. Die Art der Spinkopplung entspricht dagegen nicht ganz der Erwartung. Zwar ist im Grundzustand S = 0, allerdings lässt sich der Grundzustand nur bei tiefer Temperatur untersuchen; bei Raumtemperatur sind die beiden Spins merklich entkoppelt. Im Bereich teilweiser Spinkopplung zwischen ca. −200 und −100 °C beschreibt der Formalismus der antiferromagnetischen Kopplung korrekt den Verlauf der zunehmenden Entkopplung. Die Struktur von Kupferacetat lässt die Interpretation durchaus zu, den S=0-Grundzustand als Folge antiferromagnetischer Kopplung zu verstehen. Die magnetischen x2−y2-Orbitale sind auf die O-Atome der verbrückenden Acetato-Liganden ausgerichtet und es ergibt sich ein antiferromagnetischer Austauschpfad im Sinne der Goodenough-Kanamori-Regeln, der im folgenden Bild für einen der vier Acetato-Liganden formuliert ist. Die übrigen drei Liganden lassen sich auf die gleiche Weise behandeln, so dass durch das Zusammenwirkung der vier Austauschpfade die hohe Kopplungskonstante von J = −294 cm−1 plausibel wird. (Die x-Richtung ist in der folgenden Abbildung senkrecht zur Zeichenebene gewählt, z verläuft entlang der Cu-Cu-Achse; man beachte, dass die px-Orbitale an den O-Atomen nichtbindende Wechselwirkungen mit den x2−y2-Orbitalen der Kupferatome aufweisen, dass also nicht die delokalisierte π-Bindung des Carboxylato-Liganden für die antiferromagnetische Kopplung verantwortlich ist; die üblicherweise formulierte sp2-Hybridisierung am Carboxylat-C-Atom wurde für das Kopplungsschema aufgehoben, ferner wurden die py- und pz-Orbitale der O-Atome in geeigneter Weise linearkombiniert.)
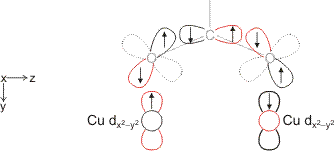
Ist im Kupfer(II)-acetat nun eine δ-Bindung oder antiferromagnetische Spinkopplung für den S=0-Zustand verantwortlich. Das Schema suggeriert den derzeitigen Stand der Diskussion: Werden die Orbitale in der aus quantenchemischen Rechnungen erhaltenen Ausdehnung dargestellt, so ergibt sich bei dem schon recht großen Abstand der Kupferzentren keine nennenswerte direkte Überlappung der beiden magnetischen Orbitale, so dass die Übertragung der Spininformation auf dem abgebildeten Superaustauschpfad den bei weitem größeren Anteil an der beobachteten Spinkopplung haben dürfte.
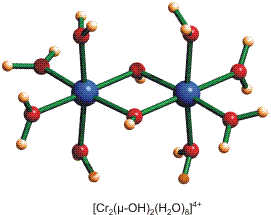
Der Frage „Bindung oder antiferromagnetische Kopplung?“ kann man sich besonders gut nähern, wenn Spezies in die Diskussion eingeschlossen werden, bei denen üblicherweise eine Bindung überhaupt nicht diskutiert wird. Ein Beispiel sind mehrkernige, oxido-verbrückte Chrom(III)-Spezies. Im folgenden soll ein Kation, in dem zwei Chrom-Atome in einem Abstand von ca. 3 Å vorliegen, auf mögliche Austauschpfade untersucht werden, um abschließend die Frage nach einer Cr-Cr-Bindung zu klären. Aufgrund der kinetischen Inertheit von Chrom(III)-Zentren gegenüber Ligandenaustauschreaktionen können hier Zwischenstufen auf dem Weg [Cr(H2O)6]3+ → Cr(OH)3 isoliert werden, die bei einem Zentralmetall wie Eisen oder Aluminium bislang nicht gefasst wurden. Ein Beispiel ist das [Cr2(OH)2(H2O)8]4+-Ion, für das eine Strukturanalyse vorliegt (Abb. auch als pdf):
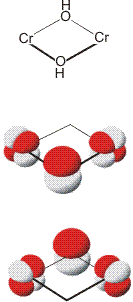
Die Lage der spintragenden Orbitale im Raum ergibt sich aus der Struktur und dem Kristallfeldmodell. Die oben erwähnten Goodenough-Kanamori(-Anderson)-Regeln eignen sich zur Voraussage der zu erwartenden Spinkopplung. (1) Antiferromagnetische Kopplung ist danach auf Pfaden zu erwarten, entlang denen ein nenneswertes Überlappungsintegral resultiert. (2) Ferromagnetische Kopplung ist dagegen zu erwarten, wenn das Überlappungsintegral 0 oder nahe 0 ist, wenn also orthogonale Orbitale in räumliche Nähe geraten. Liegen beide Pfade vor, dominiert die antiferromagnetische Kopplung.
Im folgenden Schema sind antiferromagnetische Pfade gezeigt. Die Überlappungsintegrale für die einzelnen Wechselwirkungen werden nicht so groß sein wie bei den Kupfer(II)-Beispielen. Während dort Wechselwirkungen mit lokaler σ-Symmetrie charakteristisch waren, ergeben sich bei Chrom(III) π-Bindungen:
Ferromagnetische Austauschpfade verlaufen über orthogonale Orbitale (Blick auf die Cr2(μ-OH)2-Ebene, definiert als xy-Ebene; Abbildungen auch als pdf):
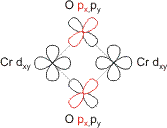
Beim ferromagnetischen Spinkopplungspfad fällt auf, dass die einfach besetzten Chrom-Orbitale in der passenden Symmetrie vorliegen, um eine Cr-Cr-Bindung aufzubauen. Diese Möglichkeit wird in der Literatur jedoch nicht erwogen. Die Ursache deutet auf einen wesentlichen Unterschied zwischen Metallen der ersten Übergangsreihe in höherer Oxidationsstufe und Metallen der beiden folgenden Übergangsreihen hin: Bei den 3d-Elementen reichen die d-Orbitale nicht wirksam in den Raum hinaus, so dass zum Beispiel in der gezeigten Anordnung keine Überlappung wirksam wird. Aus demselben Grund wurde bereits bei Kupfer(II)-acetat einer einzelnen δ-Bindung keine Bedeutung beigemessen. Chrom(II)-acetat mit der Cr-Cr-Vierfachbindung erscheint demnach als bemerkenswerte Ausnahme, man beachte aber die Argumente dafür, dass die Cr-Cr-Bindung nur schwach ist [crcr1]. Mit dem Regelfall befasst sich die folgende Lehreinheit. Die geringe Orbitalausdehnung bei Chrom(III), wo die Position in der ersten Übergangsreihe mit einer hohen Oxidationsstufe zusammentrifft, beeinflusst auch die Größe der magnetischen Kopplung. Die Goodenough-Kanamori-Regeln sagen aufgrund der konkurrierenden Austauschpfade voraus, dass die dann dominante antiferromagnetische Kopplung vorherrschen sollte. Dies wird in der Tat gefunden, allerdings ist aufgrund der eher geringen Überlappung der Sauerstofforbitale mit den kontrahierten Chrom-Orbitalen die Kopplungskonstante klein.
In einer aktuellen Arbeit wird über die erste Cr-Cr-Fünffachbindung berichtet [crcr2]. Der Cr-Cr-Abstand beträgt 1.8351(4) Å.
A. R. Sadique, M. J. Heeg, C. H. Winter: A Weak, Short Metal-Metal Bond in a Chromium(II) Amidinate Complex. J. Am. Chem. Soc. 2003, 125, 7774–7775 [crcr1].
T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long, P. P. Power: Synthesis of a Stable Compound with Fivefold Bonding Between Two Chromium(I) Centers. Science 2005, 310, 844–847 [crcr2].
Das Fazit der Lehreinheit lautet, dass die d-Orbitale bei den Elementen der zweiten und dritten Übergangsreihe viel deutlicher den Charakter von Valenzorbitalen haben als bei den 3d-Elementen. Bei den Elementen der ersten Übergangsreihe sind die d-Orbitale näher am Atomrumpf lokalisiert und zeigen – umso mehr, wenn eine hohe Oxidationsstufe vorliegt – geringe Überlappung mit den Orbitalen der Bindungspartner. Paramagnetische, eventuell antiferromagnetisch gekoppelte, seltener ferromagnetisch gekoppelte Grundzustände sind daher bei vielen Komplexen mit 3d-Elementen die Regel, während 4d- und 5d-Metalle in homologen Verbindungen Metall-Metall-Bindungen aufbauen. Lanthanoide verhalten sich in dieser Hinsicht noch extremer als die Metalle der ersten Übergangsreihe. Hier stehen die teilweise besetzten f-Orbitale für eine Orbitalüberlappung nicht zur Verfügung, nicht einmal eine merkliche Kristallfeldaufspaltung trägt zur Chemie dieser Elemente bei.
Das Prinzip findet sich zum Beispiel bei Molybdän(V). In wässrig-saurer Lösung liegt eine kationische Spezies der Summenformel MoO2(H2O)3+ vor. In der Formulierung als Einkernkomplex läge ein d1-Zentrum vor – eine Situation, die bei den 3d-Elementen wohlbekannt ist, man denke an das Hexaaqua-titan(III)-Ion [Ti(H2O)6]3+ oder an das hydratisierte Pentaaqua-oxido-vanadium(IV)-Ion („Vanadyl“-Ion) [VO(H2O)5]2+. Mit dem 5d-Element Molybdän jedoch dimerisieren die hypothetischen d1-Radikale und es entsteht eine Mo-Mo-Einfachbindung:
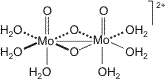
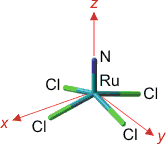
Bei der Diskussion der Mo-Mo-Einfachbindung führt das Kristallfeldmodell zu einer plausiblen Deutung der Bindung als einer Wechselwirkung mit lokaler σ-Symmetrie. Sind die Liganden sehr verschieden oder ist die Geometrie des Komplexes ungewöhnlicher, fällt es schwerer, die metallständigen Orbitale hinsichtlich ihrer energetischen Abfolgee zu ordnen. Ein Beispiel ist das [RuVINCl4]−-Ion, ein d2-Komplex, der aus RuO4, HCl und Azid zugänglich ist. Die Strukturanalyse zeigt quadratisch-pyramidalen Aufbau. Der Ru-N-Abstand ist mit 1.58 Å recht kurz; die Ru-Cl-Abstände betragen im Mittel 2.31 Å, der mittlere N-Ru-Cl-Winkel ist mit 105° stumpf. Ein erstes und schnell zugängliches Bild über die Grenzorbitale vermittelt eine DFT-Rechnung mit kleinem Basissatz (vgl. eine Gaussian-Eingabedatei; wenn Sie die Rechnung nachvollziehen wollen, ändern Sie die Datei-Extension in „gjf“). Die Koordinaten für die Rechnung sind wie folgt gewählt:
Programme wie GaussView helfen bei der Darstellung der Orbitale. Das Ergebnis ist in der folgenden Abbildung dargestellt. Die Zahlen bei den Orbitalen ist die Laufnummer in der Gaussian-Ausgabedatei. Der energetische Abstand der Orbitale ist maßstabsgerecht dargestellt; der HOMO-LUMO-Abstand ergibt sich bei der Rechnung zu ca. 0.12 Hartree, also ca. 300 kJ mol−1. Beachten Sie aber, dass vor allem errechnete Orbitalenergien unbesetzter („virtueller“) Orbitale keine sonderlich ernst zu nehmende Größe darstellen. Sie sollten also nicht in der Art schlussfolgern: 300 kJ mol−1 entspricht ca. 400 nm, also sollte das Ion bei der Anregung eines HOMO-Elektrons in das LUMO blaues Licht absorbieren und das wegen des Ligand-zu-Metall-charge-transfer-Übergangs mit hoher Extinktion, es sollte also kräftig gelb sein (es ist kräftig orange) – alles Zufall. (Bild auch als pdf.).
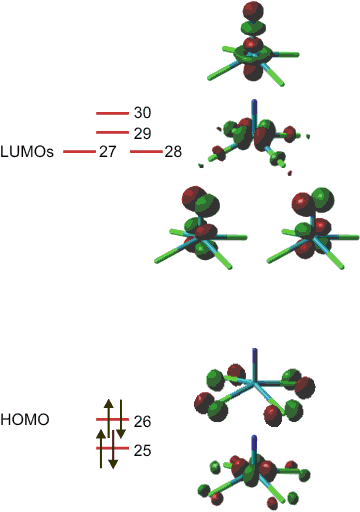
Wir diskutieren die folgenden Aspekte:
Im Grenzorbitalbereich finden sich nicht nur die erwarteten 5 Valenzorbitale des Metalls. Das HOMO ist vielmehr eine antibindende Kombination freier Elektronenpaare der Chlorido-Liganden.
Alle Wechselwirkungen zwischen Stickstoff und Ruthenium weisen im bindenden und im dargestellten antibindenden Bereich eine etwa gleich große Orbitalbeteiligung beider Atome auf – die Wechselwirkung ist kovalent.
Das Metall liegt im low-spin-Zustand vor, das freie xy-Elektronenpaar am Metall entzieht sich einer VSEPR-artigen Behandlung.
Fazit: dn-Zustände sind in der zweiten und dritten Übergangsreihe entweder Metall-Metall-bindend, oder sie führen zur low-spin-Konfiguration.
Ein weiteres Beispiel für die d2-Konfiguration, nun aber Metall-Metall-Bindungen verursachend. Die Bindungsordnung zwischen den Molybdän(IV)-Atomen im dargestellten Tetrakation ist 1.
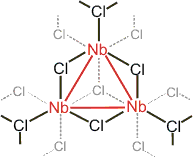
Die Tendenz zur Bildung von Metall-Metall-Bindungen wird bei den 4d- und 5d-Elementen herangezogen, um ungewöhnliche Eigenschaften zu deuten. Leitfähigkeitsmessungen zeigen zum Beispiel, dass in Kristallen von Nb3Cl8 1 bewegliches Elektron pro Formeleinheit vorliegt. In der Kristallstruktur liegen Nb3-Fragmente in Oktaederlücken einer dichtesten Chlorid-Packung vor. Die Ladung des Nb3-Fragments ist 8+, die formale Oxidationsstufe der Metallatome ist 8/3 entsprechend einer d-Elektronenzahl von 5 − 8/3 = 7/3. Werden nun jeweils 6/3 = 2 d-Elektronen für jeweils zwei Metall-Metall-Bindungen pro Niob-Atom verwendet, so bleibt 1/3 Elektron pro Niob-Atom, also 3 × 1/3 = 1 Elektron pro Formeleinheit übrig – passend zum Experiment:
Ein direkter Vergleich der d4-Zentren CrII und dem schweren Homologen MoII zeigt die Unterschiede zwischen der ersten und zweiten Übergangsreihe in besonders instruktiver Weise. Die Diskussion um Chrom(II)-acetat hat als Regel ergeben, dass Metall-Metall-Bindungen in den Strukturen sehr auffallend sind, aber hinsichtlich der Bindungsenergie nicht dominant sind. Ohne passgenau die Cr2-Einheit unterstützende Brückenliganden wird mit einer CrII-CrII-Bindung daher eher nicht zu rechnen sein.
An dieser Stelle gelingt der Bezug zur Festkörperchemie: Welcher Aufbau lässt sich aufgrund dieser Gesetzmäßigkeiten für Chrom(II)-chlorid ableiten? Welche Unterschiede sind zu erwarten, wenn anstelle von Chrom dessen schweres Homologes eingesetzt wird? Bei Molybdän(II)-chlorid treten nämlich die deutlich stärkeren MoII-MoII-Bindungen beim Aufbau des Festkörpers hervor.
Zurück zur Koordinationschemie: Welcher Aufbau darf für das Reaktionsprodukt von MoCl2 und 2 Cl− erwartet werden? Sind in ReCl3 = 1/3 Re3Cl9 Re-Re-Bindungen möglich?
Der Schwerpunkt des Kapitels lag auf Beispielen, bei welchen die Ausbildung von Metall-Metall-Bindungen als charakteristisches Verhalten der d-Elektronen von 4d- und 5d-Elementen auftrat. Generell ist diese Möglichkeit vor allem bei den frühen Übergangsmetallen zu finden. Verbindungen später, elektronenreicherer Metalle der zweiten und dritten Übergangsreihe sind durch low-spin-Zustände charakterisiert, die bei Bedarf von Metall-Metall-Bindungen begleitet werden. Das Prinzip wurde bereits bei [RuNCl4]− sichtbar. Als weiteres Beispiel werden Aufbau und Rh-Rh-Bindungsordnung in [Rh2(H2O)2(OAc)4] diskutiert.
High-spin-Cobalt(II) illustriert besonders anschaulich das Prinzip, dass die d-Elektronen der 3d-Elemente nur begrenzt die Chemie dieser Zentralmetalle mitbestimmen, sich also wie Valenzelektronen verhalten. Metall-Metall-Bindungen werden nicht beobachtet, darüber hinaus bedingt die ungefähr gleiche Ligandfeldstabilisierungsenergie bei verschiedenen Koordinationsgeometrien, dass Kristallfeldeffekte kaum wahrnehmbar sind.
Dies alles gilt jedoch nur im high-spin-Fall. Starkfeldliganden, die eine d7-low-spin-Anordnung verursachen können, verändern dieses Bild völlig. [Co(CN)4]2− ist quadratisch planar, [Co(CN)5]3− neigt zur Dimerisierung zu [Co2(CN)10]6− unter Aufbau einer Co-Co-Bindung, ein dem [Fe(CN)6]4− entsprechendes [Co(CN)6]4− ist nicht bekannt. Vor allem die Bildung einer Metall-Metall-σ-Bindung bei der Dimerisierung rückt das Cobalt(II) in die Nähe seiner schweren Homologen. Völlig analog bildet Rhodium(II) eine Rh-Rh-σ-Bindung im oben erwähnten [Rh2(H2O)2(OAc)4]. Der Unterschied zwischen Cyano- und Halogenido-Liganden wird im Orbitalschema deutlich. Halogeno-Liganden sind σ- und π-Donoren. Cyano-Liganden sind bessere σ-Donoren, deren HOMO, das 3σ-Orbital, wirksam auf das Zentralmetall zuweist. Das HOMO−1, das 1π-Orbital, ist dagegen auf das elektronegativere N-Atom ausgerichtet, so dass dessen Überlappung mit dem Zentralmetall schwach ist (vgl. die analoge Situation bei den Grenzorbitalen des Carbonyl-Liganden). Cyanid ist also aufgrund seines Aufbaus aus einem elektronegativen N- und einem weniger elektronegativen C-Atom ein Ligand, dessen σ-Donoreigenschaft verstärkt und dessen π-Donoreigenschaft geschwächt ist. Beides trägt zur Stellung des Cyano-Liganden in der spektrochemischen Reihe bei. Liegt ein Zentralmetall in niedriger Oxidationsstufe vor, kommt ein weiterer Aspekt hinzu, der in der Literatur jedoch kontrovers behandelt wird. Das LUMO, das 2π-Orbital, ist ebenfalls deutlich auf das Zentralmetall ausgerichtet. Dessen Symmetrie erlaubt eine Überlappung mit lokaler π-Symmetrie mit geeigneten d-Orbitalen des Zentralmetalls, bei einem oktaedrischen Komplex mit den t2g-Orbitalen. Sind diese besetzt, wird eine „Rückbindung“ erhalten. Das Metall wirkt hierbei als Lewis-Base, der Ligand als Säure. In der Summe zeigt sich Cyanid als starke σ-Base, als schwache π-Base und eventuell als π-Säure, wenn geeignete besetzte Orbitale am Zentralmetall zur Verfügung stehen. Unter welchen Umständen eine π-Rückbindung einen merklichen Beitrag hat, ist Gegenstand der Diskussion. Aus Röntgenspektren wurde kürzlich abgeleitet, dass selbst low-spin-Eisen(II) im Hexacyanoferrat(II) trotz seiner recht hohen Oxidationsstufe und seiner folglich eher stärker kontrahierten 3d-Orbitale eine „nennenswerte“ Rückbindung aufbauen [cyano1].
Die bei den Cyanocobaltaten(II) gefundenen Verhältnisse zumindest lassen sich zwanglos mit der starken σ-Basizität der Cyano-Liganden allein erklären. In einem oktaedrischen [Co(CN)6]4− wäre die Aufspaltung zwischen eg und eg* groß, eg* wäre stark antibindend. In dieses Schema sind 7 + 6 × 2 = 19 Elektronen einzufüllen, 1 Elektron würde sich also – anders als im [Fe(CN)6]4−-Ion – im stark antibindenden Zustand wiederfinden. Komplexe mit stark σ-basischen Liganden, zu denen zum Beispiel auch Hydrido- und Alkyl-Liganden zählen, beachten daher eine Obergrenze von 18 Elektronen. Im Fall des [Co(CN)4]2−-Ions führt die gleiche Betrachtung zur quadratisch-planaren Ligandanordnung anstelle der tetraedrischen. Auch ist es günstig, bei starkem Feld das stark antibindende x2−y2-Orbital nicht zu besetzen. Bei tetraedrischer Anordnung gibt es dagegen kein einzelnes unstabiles Orbital.
Die Betrachtung von Cyanid als σ-Donor, aber nicht zwangsweise auch als π-Akzeptor, relativiert die Fassungslosigkeit, mit der die Autoren einer aktuellen Publikation auf den für sie unerwarteten Ausgang des Versuchs der Synthese von [CrII(CN)6]4− reagieren [cyano2].
Die hohe σ-Basizität unterscheidet den Cyano-Liganden von isosteren Teilchen wie CO und NO+. Man vergleiche hierzu die Stabilitäten von HCN und HCO+. Trotz der formalen Gemeinsamkeiten unterscheidet sich die Carbonyl-Komplexchemie erheblich von der Cyanid-Metall-Chemie. Stabile Cyano-Komplexe von Metallen mit positiver Oxidationsstufe gibt es in großer Zahl, nicht jedoch die analogen Carbonylkomplexe. Man vergleiche [Cu(CN)4]3− und einen der unbeständigen Carbonyl-kupfer(I)-Komplexe wie [Cu(NH3)3(CO)]+ oder Cu(CO)Cl. Besonders illustrativ ist der Vergleich zwischen [Fe(CN)6]4− und [Fe(CO)6]2+. Das Anion des gelben Blutlaugensalzes ist seit fast 200 Jahren bekannt; es ist so stabil, dass es ungiftig ist. Das Hexa(carbonyl)eisen(II)-Ion wurde dagegen erst kürzlich von Willner und Aubke in supersauren Medien hergestellt, also in Abwesenheit aller konkurrierender Liganden. Stabile Carbonylmetall-Komplexe sind dagegen unter Bedingungen bekannt, die die höhere π-Acidität des Kohlenmonoxids ausnutzen, also mit Metallen in niedriger Oxidationsstufe, die eine hinreichend große Metallbasizität aufbringen, um die M-CO-Rückbindung zu stärken. Komplexe mit dem NO+-Ligand setzen diesen Trend fort. Beständige Nitrosyl-Komplexe sind daher nur von elektronenreichen Metallen bekannt.
Um diese Prinzipien verständlich zu machen, werden die folgenden Eisen-Komplexionen verglichen: [Fe(CO)3NO]−, [Fe(CN)5NO]2− und [Fe(H2O)5NO]2+, dem farbgebenden Komplex des „braunen Rings“. Ziel ist, die Bedeutung der π-Rückbindung, die bei der Behandlung von Cyano-Komplexen unscharf geblieben war, besser zu verstehen. Alle drei Nitrosyl-Eisen-Komplexe haben eine Gemeinsamkeit: das Fe-N-O-Fragment ist linear. Dies legt in einem einfachen VB-Bild nahe, dass der Ligand als NO+ vorliegt, isoster mit CO und CN−.
Der Standardfall des beständigen Nitrosylkomplexes mit einem fest gebundenen NO-Ligand ist im [Fe(CO)3NO]−-Ion realisiert. Die Reaktionsbedingungen für die Synthese wässriger Lösungen der intensiv gelben Alkali-tricarbonyl-nitrosylferrate(1−) zeigen schon, dass das Komplexanion keine unbeständige Spezies ist. So wird eine wässrige, schwach alkalische Alkalinitrit-Lösung mit Pentacarbonyl-eisen(0) solange am Rückfluss zum Sieden erhitzt, bis kein [Fe(CO)5] – eine mit Wasser nicht mischbare farblose Flüssigkeit – im Rücklauf des Kühlers mehr erkennbar ist:
[Fe(CO)5] + NO2− + Ca(OH)2 → [Fe(CO)3NO]− + CO + H2O + CaCO3
Sowohl [Fe(CO)5] als auch [Fe(CO)3NO]− sind typische Komplexe mit stark π-aciden Liganden, die der 18-e-Regel genügen. Um die Elektronenbilanz festzustellen, wird üblicherweise (1) aus der Ladung des Komplexes und der (von der IUPAC festgesetzten) Ladung der Liganden die Oxidationsstufe des Zentralmetallatoms bestimmt, dann (2) die Zahl der (n−1)d-Elektronen des Metalls ermittelt (der ns-Zustand wird dabei als unbesetzt angenommen) und (3) diese d-Elektronenzahl und die Zahl der (laut IUPAC-Regel) von den Liganden beigetragenen Elektronen addiert. Für [Fe(CO)5] ergibt sich wegen des Neutralliganden CO 0 als Oxidationsstufe des Eisens entsprechend 8 3d-Elektronen, hinzugezählt werden 5 × 2 = 10 Elektronen, da CO als 2e-Donor zählt, so dass sich insgesamt 18 e ergeben. Diese Zählweise steht im Einklang mit den physikalischen Eigenschaften des Pentacarbonyleisens – formale und physikalische (spektroskopische) Oxidationsstufe sind gleich. Dies ist bei Nitrosylkomplexen eher die Ausnahme. Der Nitrosylligand ist als neutraler Dreielektronen-Donor definiert. Für das [Fe(CO)3NO]−-Ion ergibt die Rechnung dann −I als Oxidationsstufe des Zentralmetalls entsprechend 9 d-Elektronen. Hinzuaddiert werden 3 × 2 = 6 Elektronen von den drei Carbonyl-Liganden und 3 Elektronen von NO. Die Summe ist wieder 18 Elektronen. Die Behandlung von NO als Neutralligand führt jedoch zu einer Reihe von Problemen bei der Deutung der physikalischen Daten. So zeigt die Strukturanalyse eine lineare Fe-N-O-Einheit, während das VSEPR-Modell eine an N gewinkelte Struktur nahelegt. Werden Abstände (in der Abbildung in Å, die Zahlenwerte sind Mittelwerte im Natriumsalz) und Winkel hinzugenommen, so ergibt sich eher die Vorstellung von einem mit CO isosteren NO+-Ligand, der den Carbonyl-Liganden hinsichtlich der π-Acidität deutlich übertrifft (Abbildung als pdf):
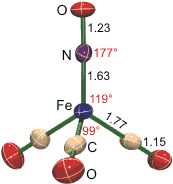
Nach dem VSEPR-Modell zeigt der größere mittlere N-Fe-C-Winkel gegenüber einem kleinen C-Fe-C-Winkel eine höhere Fe-N- im Vergleich zur Fe-C-Bindungsordnung an. Im Einklang hiermit sind die Fe-N/C-Abstände. Bezeichnend aber ist vor allem die deutliche Aufweitung des N-O-Abstands im Komplex gegenüber dem Wert für eine N-O-Dreifachbindung. Die entsprechenden Werte unterscheiden sich für CO viel weniger (C-O-Abstand in freiem Kohlenmonoxid: 1.128 Å). Mit der Ladung +1 für den Nitrosylligand ergibt sich für Eisen die physikalische Oxidationsstufe -II, die einer d10-Konfiguration entspricht, wie sie auch im isosteren Tetracarbonyl-ferrat(−II) gefunden wird. Dieses homoleptische Ferrat entsteht bei der Umsetzung von [Fe(CO)5] mit wässriger Lauge (Hiebersche Basereaktion):
[Fe(CO)5] + 4 OH− → [Fe(CO)4]2− + 2 H2O + CO32−
Der Zweielektronen-Reduktion des Eisenzentrums steht bei beiden Reaktionen die Oxidation eines Äquivalents CO zu CO2 gegenüber, das unter den basischen Reaktionsbedingungen als Carbonat anfällt. Das Natriumsalz des Tetracarbonylferrats wird als „Collmans Reagenz“ verwendet. Die höhere π-Acidität des NO+-Liganden im Vergleich mit CO lässt sich auch anhand der C-O-Schwingungsfrequenzen nachvollziehen: 1790 cm−1 bei Tetracarbonylferrat gegenüber ca. 1900 cm−1 bei Tricarbonylnitrosylferraten, bei denen der NO-Ligand mehr Metallbasizität auf sich zieht (vgl. 2143 cm−1 bei freiem CO).
Die Vorstellung, dass eine lineare M-N-O-Einheit einen CO-analogen NO+-Liganden anzeigt, ist in Lehrbüchern verbreitet, aber falsch. Der von der Nitratprobe bekannte braune Ring, dessen Chromophor bei der Nitritprobe im gesamten Probevolumen entsteht, wird durch den Pentaaqua-nitrosyl-eisen(II)-Komplex [Fe(H2O)5NO]2+ hervorgerufen. Für dieses Ion liegt keine Strukturanalyse vor, es herrscht jedoch Einigkeit, dass die spektroskopischen Daten und die Ergebnisse von Rechnungen ein lineares Fe-N-O-Fragment hinreichend belegen. Der Grundzustand ist ein Quartett (S = 3/2). In vielen Lehrbüchern wird der Nitrosyl-Ligand daher als NO+ eingestuft, der an ein high-spin-d7-Eisen(I)-Zentralatom gebunden ist. Die physikalische Oxidationsstufe des formalen Eisen(II)-Zentrums wäre demnach +I. Die Unterschiede der beiden Nitrosyleisen-Komplexe [Fe(CO)3NO]− und [Fe(H2O)5NO]2+ sind gravierend. Während das Ferrat(−II) thermisch belastbar ist und hohe Reaktivität nur gegenüber Oxidationsmitteln zeigt, ist das Ion des braunen Rings nur wenig stabil und zerfällt leicht wieder. Aktuelle Rechnungen zeigen, dass das Pentaaqua-nitrosyl-eisen-Ion eher als FeII-NO• oder als FeIII-NO−-Komplex zu verstehen ist [brown_ring1], [brown_ring2]. Wie aber ist dann die lineare Anbindung des Nitrosyl-Liganden an das Eisenzentrum zu verstehen? Wäre gemäß dem VB-Bild zum Beispiel für den FeIII-NO−-Komplex nicht ein Fe-N-O-Winkel von ca. 120° zu erwarten? Man beachte jedoch die geringe Stabilität der Fe-NO-Bindung. Das VB-Bild steht für die Koordination eines Metallzentrums an eine Singulett-NO−-Spezies. Ein high-spin-Eisenzentrum der Oxidationsstufe +II oder +III führt jedoch nicht zu einem so drastischen Eingriff in die Elektronenstruktur des Liganden. Dieser ist isoelektronisch zu O2 und liegt wie dieses in einem Triplett-Grundzustand vor. Die nun mögliche Wechselwirkung zwischen den beiden π*-Orbitalen des Liganden und symmetrisch passenden Eisen-Orbitalen ist für eine von zwei Wechselwirkungen dargestellt; die gezeigte Spinkopplung wird meist als antiferromagnetische Kopplung verstanden:
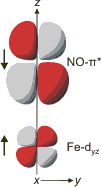
Es wird deutlich, dass sowohl Triplett-NO− als auch, in alternativer Betrachtung, das NO-Radikal linear an das Eisenzentrum binden. Aus der DFT-Rechnung in [brown_ring1] folgt ein Energieschema für die Grenzorbitale des Komplexes (Abbildungen auch als pdf). Dargestellt sind diejenigen Orbitale, die im Bereich des FeNO-Fragments lokalisiert sind. Die Nummern verweisen auf Tabelle 8 in [brown_ring1], außerdem sind die beteiligten Fe-Orbitale genannt; man beachte aber, dass hiermit ein Elektron nicht allein dem jeweiligen Orbital zugewiesen wird; die beiden besetzten β-Spin-Orbitale haben zum Beispiel jeweils zur Hälfte Metall- und NO-π*-Charakter. Die braune Farbe von [Fe(H2O)5NO]2+ geht auf Übergänge des Typs π→σ* zurück:
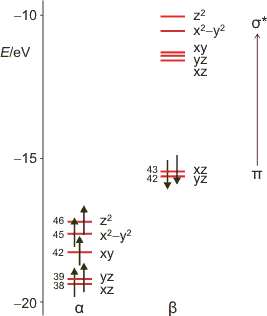
Noch ein Kommentar zur Nomenklatur: Zur Beschreibung von Nitrosyl-Metall-Komplexen ist die von Enemark und Feltham eingeführte Angabe der Elektronenzahl des MNO-Fragments allgemein üblich. Im braunen Ring liegt zum Beispiel ein {FeNO}7-Zentrum vor entsprechend 7 Elektronen als Summe der d-Elektronen des Metalls und der Elektronen in antibindenden NO-Orbitalen. Durch diese Definition umgeht man eine Festlegung auf einen speziellen Bindungsmodus.
[Fe(CO)3NO]− und [Fe(H2O)5NO]2+ erscheinen als zwei Extremfälle für die Bindung eines Nitrosylliganden. Das Eisen(−II)-Zentrum baut eine starke Rückbindung zum NO-Liganden auf und es entsteht ein Komplex, der nicht zur Dissoziation des Nitrosyl-Liganden neigt. Das Lewis-saure high-spin-Eisen(+II)-Zentrum dagegen ist nicht in der Lage, eine nennenswerte Rückbindung aufzubauen, umgekehrt ist der Nitrosyl-Ligand kein besonders guter Donor – heraus kommt ein zersetzlicher Komplex.
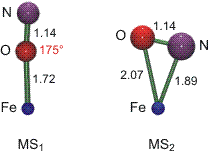
Die für eine starke Rückbindung nicht förderliche Oxidationsstufe +II liegt jedoch auch im lange bekannten Nitroprussid-Anion vor. Dessen Natriumsalz, Natrium-pentacyano-nitrosyl-ferrat(III)-Dihydrat, Na2[Fe(CN)5NO] · 2 H2O, ist ein geläufiges Medikament, das zum Beispiel bei Operationen nach Bedarf infundiert wird um den Blutdruck des Patienten schnell und wirksam zu senken. Nach der Entdeckung der Hormonwirkung von NO wurde erkannt, dass das Ferrat-Ion unter physiologischen Bedingungen NO abspaltet, worauf die Wirkung beruht. NO ist also offensichtlich auch an diesem Eisen(II)-Zentrum weniger fest gebunden als es die Strukturanalyse nahelegt:
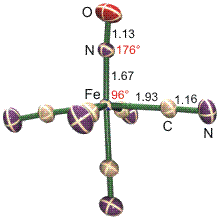
Die Strukturparameter des {FeNO}6-Zentrums im Natrium-nitroprussid zeigen wieder ein lineares Fe-N-O-Fragment. Die Fe-N-Bindung ist recht kurz, die gegenüber dem Carbonylferrat deutlich geschwächte Rückbindung zeigt sich jedoch am stark verkürzten N-O-Abstand. Um zu verstehen, dass die Fe-N-Bindung bei weitem nicht so schwach ist wie beim Aqua-nitrosyl-Komplex, ist die low-spin-Konfiguration des Cyanokomplexes zu beachten. Die sich andeutende Mittelstellung der Fe-N-Bindung – nicht so stabil wie bei einem Eisen(−II)-Zentrum, nicht so schwach wie bei high-spin-Eisen(II) – wird am besten deutlich, wenn eine aufsehenerregende Eigenschaft des Natrium-nitroprussids in die Diskussion einbezogen wird (Hauser, 1977). Beim Bestrahlen von SNP-Kristallen (SNP = sodium nitroprusside) mit grünem Laserlicht bei tiefer Temperatur wird ein metastabiler Zustand (MS2) erhalten, der einer Population von etwa der Hälfte aller Ferrat-Ionen entspricht. Beim Aufwärmen über 165 K geht MS2 in einen weiteren metastabilen Zustand MS1 über. Beide metastabile Zustände haben in der Kälte eine beliebige Lebensdauer. Der Grundzustand kann entweder durch Aufwärmen auf Raumtemperatur oder durch Bestrahlen mit rotem Laserlicht wieder erreicht werden. Die ungewöhnliche Lebensdauer deutet an, dass es sich bei den metastabilen Zuständen nicht um elektronische Anregungen handelt, sondern dass eine strukturelle Veränderung stattfindet. Diese wurde 1997 erstmals durch Röntgenbeugung nachgewiesen. Aktuelle Rechnungen unterstützen den Befund, dass in MS2 die Nitrosylgruppe eine Art π-Donor-Bindungsmodus einnimmt, der in die Isonitrosyl-Struktur MS1 relaxiert. Im Einklang mit der Arbeitshypothese, dass eine Fe-N-Rückbindung bei Eisen(II)-Zentren nicht nenneswert ist, unterscheiden sich die N-O-Abstände in den drei Formen kaum:
Eine Übersicht zu der an SNP beschriebenen photoinduzierten Bindungsisomerie (engl. photoinduced linkage isomerism) gibt [pli1].
H.-Y. Cheng, S. Chang, P.-Y. Tsai: On the “Brown-Ring” Reaction Product via Density-Functional Theory. J. Phys. Chem. A 2004, 108, 358–361 [brown_ring1].
A. Wanat, T. Schneppensieper, G. Stochel, R. van Eldik, E. Bill, K. Wieghardt: Kinetics, Mechanism, and Spectroscopy of the Reversible Binding of Nitric Oxide to Aquated Iron(II). An Undergraduate Text Book Reaction Revisited. Inorg. Chem. 2002, 41, 4–10 [brown_ring2].
A. S. Vinogradov, A. B. Preobrajenski, A. Knop-Gericke, S. L. Molodtsova, S. A. Krasnikov , S. V. Nekipelov, R. Szargan, M. Hävecker, R. Schlögl: Observation of back-donation in 3d metal cyanide complexes through N K absorption spectra. J. Electron Spectr. Rel. Phen. 2001, 114–116, 813–818 [cyano1].
K. J. Nelson, I. D. Giles, W. W. Shum, A. M. Arif, J. S. Miller: The Myth of Cyanide Always Being a Strong-Field Ligand: Synthesis and Structural Characterization of Homoleptic S = 2 Pentacyanochromate(II), [CrII(CN)5]3−, and Nonacyanodichromate(II), [CrII2(CN)9]5−. Angew. Chem. 2005, 117, 3189–3192 [cyano2].
P. Coppens, I. Novozhilova, A. Kovalevsky: Photoinduced Linkage Isomers of Transition-Metal Nitrosyl Compounds and Related Complexes. Chem. Rev. 2002, 102, 861–883 [pli1].
Das folgende qualitative MO-Schema zeigt den Bereich der Grenzorbitale eines oktaedrischen d6-Komplexes (man beachte, dass das Schema für NO hyothetisch ist, so etwas wie [Ti(NO)6]4+ ist unbekannt; Schema auch als pdf):
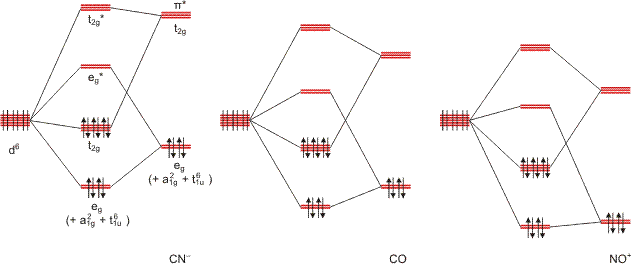
Die 18-Elektronen-Regel gilt dann, wenn die xy-, xz- und yz-Orbitale (t2g unter Oh) bindend sind. Der in der Abbildung skizzierte Unterschied zwischen Cyano-, Carbonyl- und Nitrosyl-Liganden ergibt sich bei konstanter Energie der Metallorbitale. Vor allem die in der Literatur umstrittene Charakteristik des Cyano-Liganden – ist er im wesentlichen nur σ-Donor- oder auch π-Akzeptorligand? – wird durch eine solche Darstellung transparent. Liegen Metalle in höherer Oxidationsstufe vor, sind die Metallorbitale stabilisiert und energetisch weit von den Ligand-π*-Orbitalen entfernt – eine Rückbindung wird erschwert sein. Bei Erniedrigung der positiven Ladung des Metalls nimmt die Orbitalenergie zu, die Metallorbitale bewegen sich also entlang der Energieachse auf die gleiche Weise, wie es in der Abbildung dem Gang vom Cyano- zum Nitrosylliganden entspricht. Zumindest bei Cyanokomplexen von Metallen in niedriger Oxidationsstufe ist demnach mit einer deutlichen Beteiligung von Rückbindung zu rechnen. Die signifikante Zunahme von Rückbindung zeigt sich besonders deutlich in isoelektronischen Reihen. Man verfolge als Maß für die zunehmende Rückbindung in der Reihe [Fe(CN)5NO]2−, [Mn(CN)5NO]3−, [Cr(CN)5NO]4− die NO-Valenzschwingungsfrequenzen von 1939 cm−1 (Fe), 1725 cm−1 (Mn) und 1515 cm−1 (Cr). Übung: Man gebe die formale und die spektroskopische Oxidationsstufe der Metallzentren an. Zum Vergleich: Die N-O-Valenzschwingungsfrequenz beträgt im [Fe(CO)3NO]−-Ion 1640 cm−1.
Den in der Abbildung gezeigten Fällen ist gemeinsam, dass HOMO und LUMO metallständige Orbitale sind. Die elektronische Anregung mit der niedrigsten Energie wird daher ein Übergang zwischen den d-Orbitalen des Metalls sein. Die besonderen Eigenschaften einiger Nitrosylkomplexe, nach Bestrahlung langlebige metastabile Zustände unter Rotation des Nitrosylliganden zu entwickeln, wird so nicht plausibel. Rechnungen am Nitroprussid-Anion zeigen jedoch, dass durch die Substitution eines von sechs Cyano-Liganden eines Hexacyanoferrat(II)-Ions durch NO+ ein antibindendes NO-π*-Orbital unter den eg*-Zustand eingeschoben ist (P. Boulet, M. Buchs, H. Chermette, C. Daul, F. Gilardoni, F. Rogemond, C. W. Schläpfer, J. Weber, J. Phys. Chem. A 2001, 105, 8991–8998; die Orbitalenergien sind für ein Anion unerwartet niedrig, die Autoren geben aber keine Einzelheiten an, vermutlich haben sie ein isotropes positives Feld zugeschaltet):
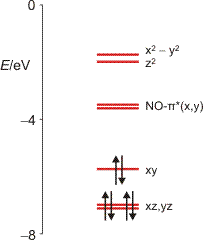
Die optische Anregung ist als Folge der NO-π*-Lage ein MLCT-Übergang (MLCT = metal to ligand charge transfer), der die Fe-N-Bindung schwächt und so die Rotation einleitet. Die Bildung dieser metastabilen Zustände ist an den Festkörper gebunden, die Rotation findet nach der Bindungsschwächung in einem begrenzten Hohlraum der Kristallstruktur statt. In Lösung wäre die Abdissoziation des Liganden zu erwarten – siehe das oben Gesagte zur medizinischen Anwendung von SNP.
Bei Komplexen, welche die 18-Elektronenregel beachten, ist die Elektronendonorfähigkeit des Metallzentrums mit der Ausbildung von Rückbindungen oft noch nicht erschöpft. Solche Zentren weisen Metallbasizität auf, sie leiten sich oft von Fragmenten ab, die isolobal zu Nichtmetall-Fragmenten sind und deren recht hohe Elektronegativität teilen.
Pentacarbonyleisen, eine farblose Flüssigkeit, die sich durch die Umsetzung von feinverteiltem Eisen mit CO herstellen lässt, ist ein 18-Elektronen-Komplex (8 e von Fe(0) + 5 × 2 e von 5 CO = 18 e). Fe(CO)5 reagiert mit zahlreichen Nucleophilen. Die dabei entstehenden Komplexe eignen sich gut, das Phänomen der Metallbasizität zu untersuchen. Im folgenden Schema ist zuerst die Umsetzung mit verschiedenen Nucleophilen angegeben, anschließend werden die neuen Komplexe mit einem Elektrophil umgesetzt, das als Sonde zur Detektion von Basizität dient. Im Schema bedeutet eine freie Bindung eine Carbonylgruppe.
Bei der Umsetzung mit Nucleophilen werden zwei typische Reaktionsmöglichkeiten beobachtet: Nucleophile wie Cyanid oder Nitrit, die gute Liganden sind oder bilden können, ersetzen einen oder mehrere Carbonyl-Liganden. Harte Nucleophile wie OH− oder Alkyle greifen das Kohlenstoffatom eines Carbonyl-Liganden an. Der bei der mit OH− ablaufenden Hieberschen Basenreaktion intermediär gebildete Hydroxycarbonyl-Ligand führt dabei durch β-Eliminierung zur Bildung von Tetracarbonyl-ferrat(−II), während mit Alkyl- oder Aryl-Anionen die stabileren Acyl-Liganden gebildet werden.
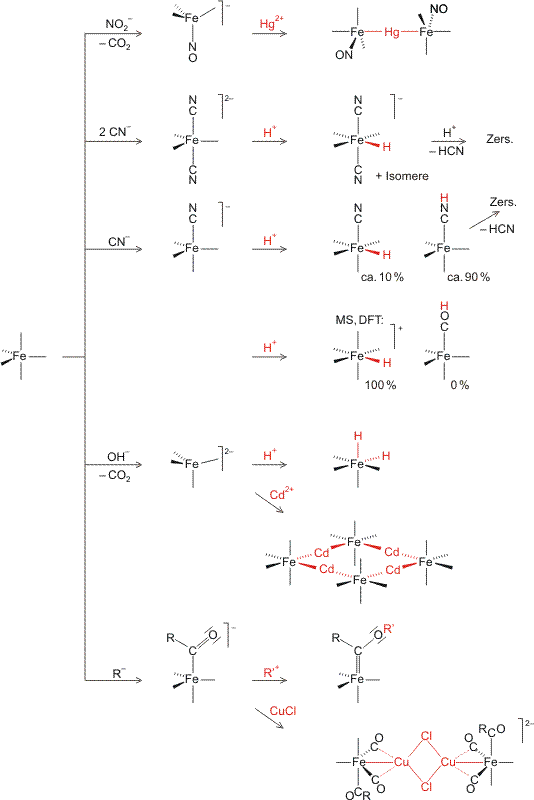
Schema auch als pdf.
In der ersten Spalte des Schemas sind die durch den Angriff eines Nucleophils gebildeten Komplexe zusammengestellt. Durch die Reaktion mit Elektrophilen lässt sich nun deren Metallbasizität untersuchen.
Im Fe(CO)5 selbst ist die Basizität des Eisenzentrums durch die Ausbildung von Rückbindungen weitgehend erschöpft. Die Stammverbindung ist protonierbar, das Produkt [HFe(CO)5]+ lässt sich jedoch nur massenspektrometrisch nachweisen. Nach DFT-Rechnungen bindet das Wasserstoffatom an das Metallzentrum und nicht an ein O-Atom eines Carbonyl-Liganden. Dieses Verhalten lässt sich auf aufschlussreiche Weise modifizieren, wenn zuerst 1, dann 2 CO- durch Cyano-Liganden ersetzt werden. Die Konkurrenz zwischen Metallbasizität und Ligandbasizität wird anhand der Cyano-carbonylferrate diskutiert.
Präparativ ist die Reaktion mit anderen Elektrophilen als dem Proton von Bedeutung. Im Schema sind drei Beispiele angegeben, bei denen durch Umsetzung eines Lewis-basischen Ferrats mit einem Lewis-sauren Metallkation Metall-Metall-Bindungen aufgebaut werden können. Ist die Basizität von Metallzentrum und basischen Ligandatomen hinreichend gleich verteilt, so lässt sich mit dem HSAB-Konzept der Ort des elektrophilen Angriffs einordnen. Ein Beispiel ist die Umsetzung von Acyl-tetracarbonyl-ferraten (1) mit Reagenzien, die Alkyl-Kationen übertragen, und (2) mit Kupfer(I)-Salzen.
Im ersten Fall wird das härter basische O-Atom des Acyl-Liganden alkyliert. Ein typisches elektrophiles Reagenz ist Trimethyloxonium-tetrafluoridoborat; die Reaktionsprodukte sind „Fischer-Carbene“, die sich von ihrerseits elektrophilen Carbenen ableiten, die auch nicht metall-gebunden im Singulett-Grundzustand vorliegen und als Donor-Akzeptor-Liganden ähnlich wie CO reagieren.
Mit CuCl hingegen wird ein weiches Elektrophil angeboten, das zum Aufbau einer Metall-Metall-Bindung neigt.
Pentacarbonyleisen spaltet beim Erhitzen CO ab. Zuerst entsteht Fe2(CO)9, dann Fe3(CO)12, das gezielter durch Oxidation von Tetracarbonylferrat(2−) erhalten werden kann. Strukturanalyse und IR-Spektren zeigen für den Zweikernkomplex im Kristall und in Lösung eine D3h-symmetrische Struktur mit 3 verbrückenden Carbonylgruppen, also ein Hexacarbonyl-tris(μ-carbonyl)-dieisen(0). Der Aufbau von Fe3(CO)12 war längere Zeit umstritten. Die erste Strukturanalyse war nicht eindeutig, und eine auf verbrückende CO-Gruppen hinweisende IR-Bande ist neben einer Hauptabsorption bei 2050 cm−1 und weiteren Banden im Bereich terminaler Carbonylliganden nur schwach bei 1870 cm−1 zu erkennen. Die seit 1993 vorliegende korrekte Strukturanalyse wird durch neuere DFT-Rechnungen bestätigt: Im Grundzustand ist die Struktur nicht D3h-symmetrisch ohne verbrückende Carbonyl-Liganden, sondern zwei der drei Eisenatome sind von zwei Carbonylgruppen überbrückt. Die Symmetrie des Moleküls ist C2v, es entsteht formal durch Ersatz einer der drei μ-CO-Liganden in Fe2(CO)9 durch ein Fe(CO)4-Fragment, das nur terminale CO-Liganden aufweist. Die D3h-symmetrische Struktur ist ca. 20–30 kJ mol−1 weniger stabil. Sie hat in Lösung wahrscheinlich Bedeutung. Darüberhinaus fluktuiert die C2v-Struktur in Lösung (1 Signal im 13C-NMR-Spektrun) und wahrscheinlich auch im Feststoff, indem die Brückenliganden ständig ihren Platz wechseln. Man beachte, dass die IR-Spektroskopie als „schnelle“ Methode terminale und verbrückende Liganden zeitaufgelöst zeigt, während die langsamere NMR-Spektroskopie den äquilibrierten Zustand wiedergibt.
Das homologe Os3(CO)12 liegt dagegen in eben dieser D3h-symmetrischen Form ohne verbrückende CO-Liganden vor. Die DFT-Rechnung weist hier Isomere mit μ-CO-Liganden als unstabil aus. Ursache ist die stärkere Kontraktion der 3d-Orbitale der Eisenzentren im Vergleich zu den 5d-Orbitalen des Osmiums. So lässt sich zeigen, dass die Bindung zu μ-CO-Gruppen die Metall-Metall-Bindung schwächen. Während der Energieverlust durch eine schwächere Metall-Metall-Bindung bei den aufgrund der geringeren Orbitalüberlappung ohnehin schwächeren Eisen-Eisen-Bindungen durch den Aufbau von mehr Fe-C-Kontakten überkompensiert wird, ist dies bei Osmium umgekehrt. Wie schon in der Koordinationschemie höherer Oxidationsstufen ergibt sich der Unterschied zwischen erster Übergangsreihe und den schweren Homologen aus der stärkeren Lokalisierung der d-Elektronen am Atomrumpf im Fall eines 3d-Elementes [cluster1].
Die Bindung von Carbonylliganden als terminale oder verbrückende Liganden ändert in der Regel nichts an der Elektronenbilanz. Ist ein verbrückender Carbonylligand nur über das C-Atom an zwei oder drei Metallatome gebunden, steht für die Hinbindung wie beim terminalen Bindungsmodus das freie Elektronenpaar am Kohlenstoffatom zur Verfügung. CO ist ein 2-Elektronen-Donor. Die üblichen Bindungsmodi verbrückender CO-Liganden sind im folgenden Schema zusammengestellt (pdf).
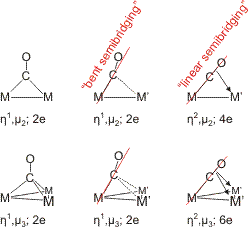
Die rechts gezeichneten Fälle des linear halbverbrückenden Modus, bei dem auch M = M' sein kann, sind selten. Sie haben besondere elektronische Verhältnisse zur Voraussetzung. Ein Beispiel für den η2, μ3-6e-Fall liegt im dreikernigen Komplex [Nb3(cp)3(CO)7] vor (Hermann, 1981). Hier ist M = M' = Nb(cp)(CO)2. Der die Bildung eines Clusters verursachende Ligandmangel wurde durch CO-Abspaltung aus dem einkernigen Carbonylkomplex [Nb(cp)(CO)4] durch Bestrahlen mit UV-Licht herbeigeführt. Ein cyclischer [{Nb(cp)(CO)n}m]-Cluster mit zwei Niob-Niob-Einfachbindungen pro Metallatom darf für n = 3 erwartet werden (siehe unten). Ein dreikerniger Cluster (m = 3) hätte dann die Zusammensetzung [Nb3(cp)3(CO)9] – was wohl zu einer sterischen Überladung führen würde. Der gefundene Cluster enthält zwei Carbonyl-Liganden weniger und trotzdem vermittelt die Struktur den Eindruck eines beachtlichen „Gedränges“: jedes Niob-Atom ist von zwei terminalen Carbonyl-Liganden koordiniert; der Cyclopentadienyl-Ligand ist wie üblich η5 gebunden. Man beachte, dass im Bild an jedem Cyclopentadienyl-Liganden der Übersichtlichkeit halber 5 H-Atome nicht eingezeichnet sind, die aber natürlich auch noch Platz brauchen.
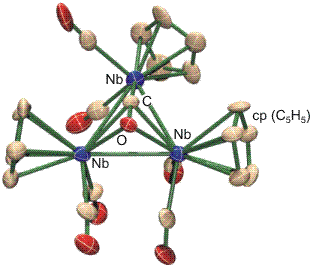
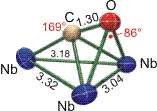
Die weiter unten zusammengefassten Elektronenabzählregeln zeigen, dass dem Cluster wegen des Verzichts auf zwei CO-Liganden vier Elektronen zur Erfüllung der 18-Elektronenregel fehlen – wenn alle CO-Liganden als 2-Elektronendonoren gezählt werden. Dass die 18-Elektronen-Regel einen hohen Stellenwert hat, wird bei einem genaueren Blick auf den μ3-CO-Liganden deutlich, der eine Reihe von Besonderheiten aufweist. Üblicherweise ist das Sauerstoffatom eines verbrückenden Carbonyl-Liganden von den Metallatomen weggeneigt (siehe oben im Schema). Aus dem im folgenden Bild dargestellten Ausschnitt aus der Struktur des Niobclusters geht jedoch hervor, dass das Fe-C-O-Fragment nicht nur entsprechend der Einstufung “linear semibridging” mit einem Winkel von 169° am C-Atom nur wenig geknickt ist, sondern das vielmehr die geringe Abwinklung darauf zurückzuführen ist, dass das O-Atom sich zwei Nb-Atomen zuneigt. Der Nb-O-Nb-Winkel von 86° (Nb-C-Nb 85°) liegt nahe am rechten Winkel – so wie man es für zwei Donorbindungen erwarten darf, die von den senkrecht aufeinander stehenden bindenden π-Orbitalen zwischen C und O ausgehen. Die C-O-Bindung wird durch diese neuen Bindungen zu zwei Niob-Atomen merklich geschwächt. Der C-O-Abstand ist mit 1.30 Å gegenüber freiem CO (1.13 Å) deutlich aufgeweitet; man vergleiche den oben angegebenen Wert von 1.15 Å im [Fe(CO)3NO]−-Ion, 1.20 Å einer C=O-Doppelbindung und 1.43 Å einer C–O-Einfachbindung. Die deutlich herabgesetzte Bindungsordnung führt zu einem für Carbonylkomplexe ungewöhnlich niedrigen Wert für die Energie der C-O-Streckschwingung von nur 1330 cm−1. Vor allem dieser letzte Wert passt zu der Interpretation, dass der verbrückende Carbonylligand als 6-Elektronen-Donor fungiert. Es ist jedoch zu beachten, dass eine 18-Elektronenbilanz auch durch 1 Nb=Nb-Doppelbindung erreicht würde. Die im Bild angegebenen Nb-Nb-Abstände zeigen, dass die Metall-Metall-Abstände tatsächlich nicht alle gleich groß sind. Da keine neueren Rechnungen an diesem Cluster vorliegen, bleibt hier Deutungsspielraum.
Dass die Aktivierung eines Carbonyl-Liganden bis zum C-O-Bindungsbruch führen kann, zeigen die beiden folgenden Beispiele: Wird Fe(CO)5 erhitzt, so entstehen unter CO-Abspaltung mehrkernige Carbonylmetall-Komplexe wie Fe2(CO)9 oder Fe3(CO)12. Wird weiter erhitzt, so werden bei ca. 200 °C Carbidocluster erhalten, in denen neben intakten Carbonyl-Liganden Carbido-Liganden, C4−, als Spaltprodukte auftreten. So lässt sich ein [Fe5C(CO)15] isolieren. Auch Cluster dieser Größe gehorchen der 18-Elektronen-Regel. Da der Umgang mit Oxidationsstufen hier sehr sperrig wäre, werden die Clusterbausteine als elektroneutral angesehen. Die bei Fe und C eingesetzten Elektronenzahlen sind also die der neutralen Atome. CO zählt wie üblich als 2-Elektronen-Donor. Man beachte, dass bei dieser summarischen Behandlung 2 Elektronen pro Metall-Metall-Bindung zu zählen sind, da erst im letzten Schritt durch die Zahl der Metallatome dividiert wird:
| [Fe5C(CO)15] | |||
|---|---|---|---|
| 5 Fe | 5 × 8 | = | 40 |
| 15 CO | 15 × 2 | = | 30 |
| 1 C | 1 × 4 | = | 4 |
| 8 Fe–Fe | 8 × 2 | = | 16 |
| Σ | = | 90 | |
| pro Fe | : 5 | = | 18 |
Weitere Beispiele für die CO-Aktivierung sind alle katalytischen Reaktionen, bei denen Kohlenmonoxid – meist zusammen mit Wasserstoff als „Synthesegas“ – als Reaktand eingesetzt wird. Zu den Reaktionen, bei denen die C-O-Bindung vollständig gebrochen wird, gehört die Fischer-Tropsch-Reaktion (FT-Synthese), die mit Cobaltkatalysatoren der folgenden Gleichung folgt:
CO + 2 H2 → (-CH2-)n/n + H2O
In allen neueren Varianten werden Eisenkatalysatoren eingesetzt, die außerdem das Wassergasgleichgewicht (CO + H2O = CO2 + H2) katalysieren. Die Gleichung für die FT-Synthese lautet dann:
2 CO + H2 → (-CH2-)n/n + CO2
Die FT-Synthese ist eine heterogen katalysierte Polymerisation von C1-Bausteinen, die sich auf einer Metalloberfläche (Eisen, Cobalt, Ruthenium) primär zu 1-Alkenen zusammenfinden. Die Kettenlänge ist steuerbar. Typischerweise werden Kohlenwasserstoffgemische angestrebt, die als Treibstoffe eingesetzt werden können. Die FT-Synthese gehört neben der Kohlehydrierung zu den Verfahren, mit deren Hilfe Erdöl durch Kohle ersetzt werden kann.
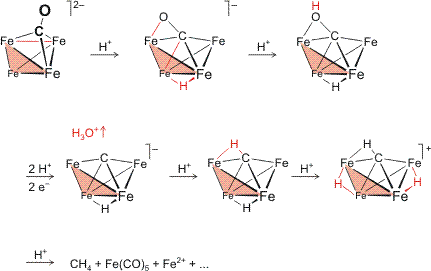
In der Literatur werden einige mechanistische Vorschläge gemacht. In einem Schlüsselexperiment konnte Brady zeigen, dass der Carbid-Methylen-Mechanismus wohl der realistischste ist. Dieser formuliert die C-O-Bindungsspaltung und die Polymerisation als zwei völlig voneinander getrennte Schritte mit sauerstofffreien C1-Bausteinen bei Kettenstart und Kettenfortpflanzung. Das folgende Schema zeigt schematisch die Einzelschritte: Adsorbiertes CO wird zu oberflächengebundenem Carbid und Oxid. Das Oxid reagiert mit adsorbiertem Wasserstoff zu Wasser und verlässt die Oberfläche. Carbid reagiert ebenfalls mit Wasserstoff, es bildet sich oberflächengebundenes Methylidin (CH) und Methylen (CH2), außerdem wird Wasserstoff als Hydrido-Ligand an die Metalloberfläche gebunden. Der Kettenstart ist die Insertion einer Methylengruppe in eine Metall-Hydrid-Bindung, es entsteht ein Methyl-Ligand. Das Kettenwachstum stellt eine Folge von Methylen-Insertionen in Metall-Alkyl-Bindungen dar, durch welche die Alkylreste verlängert werden. Der Kettenabbruch schließlich ist eine β-Wasserstoff-Eliminierung, bei der das oberflächengebundene Hydrid wiederhergestellt wird und ein 1-Alken entsteht (im technischen Prozess werden Folgeprodukte anstelle dieses reaktiven Primärproduktes erhalten). Schematisch:
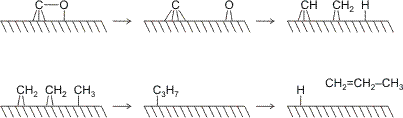
In Bradys Schlüsselexperiment wurden oberflächengebundene Methylen-Liganden durch Umsetzung einer sauberen Metalloberfläche mit Diazomethan erzeugt. Als Metalle wurden FT-Katalysatoren und Kupfer eingesetzt. Beim Erhitzen aller Methylen-belegten Oberflächen entstand Ethen durch Dimerisierung der Methylengruppen. In Anwesenheit von Wasserstoff jedoch bildeten die FT-Katalysatoren 1-Alkene – nur mit Kupfer, das nicht zur Bildung von Hydrid neigt (Kupfer ist kein Hydrierkatalysator!), blieb es auch bei Anwesenheit von Wasserstoff bei der Ethen-Bildung.
Der erste Teilschritte der FT-Synthese, die Spaltung von CO sowie die Bildung von Wasser und CHn-Spezies, ist auf molekularer Ebene nachgestellt worden (1981). Die Versuchsreihe wird in Lehrbüchern häufig zitiert, da es nur sehr wenige Beispiele gibt, bei denen so viele Einzelschritte einer Heterogenkatalyse an einem Metallcluster nachgebildet wurden. Ausgangspunkt ist ein Carbonylferrat, das sich neben anderen bei der Umsetzung höherkerniger Eisencarbonyle mit Base bildet, und zwar [Fe4(CO)13]2−. In diesem vierkernigen Cluster sind jeweils drei terminale CO-Liganden an jedem Eisenatom gebunden. Der dreizehnte Carbonyl-Ligand liegt im η1,μ3-Bindungsmodus vor, indem das C-Atom eine Dreiecksfläche des tetraedrischen Clusters überbrückt. Das Ferrat befolgt die 18-Elektronen-Regel:
| [Fe4(CO)13]2− | |||
|---|---|---|---|
| 4 Fe | 4 × 8 | = | 32 |
| 13 CO | 13 × 2 | = | 26 |
| [ ]2− | = | 2 | |
| 6 Fe–Fe | 6 × 2 | = | 12 |
| Σ | = | 72 | |
| pro Fe | : 4 | = | 18 |
Im folgenden Schema ist die Reaktionssequenz dargestellt, die nach stufenweiser Protonierung mit starker Säure (Fluor- oder Trifluormethyl-sulfonsäure) abläuft und die bei tiefer Temperatur in Einzelschritte aufgelöst werden konnte:
Das im ersten Schritt lagert sich ein Proton an den Cluster an. Dabei hat es die Wahl zwischen den weich-basischen Metallzentren und den hart-basischen O-Atomen der Carbonyl-Liganden. Unter den CO-Gruppen wiederum sollte das O-Atom des verbrückenden Liganden die höchste Basizität aufweisen, da durch Rückbindungen am meisten Ladungsdichte auf diese Carbonylgruppe übertragen sein sollte. Das Experiment zeigt, dass sich das Proton für die weich-basische Position entscheidet und sich an zwei Metallatome anlagert, die es als formaler μ2-Hydrido-Ligand verbrückt.
Die sich nun anschließende strukturelle Veränderung im Cluster spiegelt die bislang betrachteten Gesetzmäßigkeiten wider: Die 18-Elektronen-Regel wird durch die Protonierung zuerst einmal nicht berührt. Trotzdem kommt es zu einer strukturellen Veränderung, indem eine der sechs Metall-Metall-Bindungen im Produkt aufgehoben ist (im Schema ist diese im Edukt rot gezeichnet).
Eine mögliche Interpretation ergibt sich aus der vergleichenden Betrachtung der Bindungsverhältnisse in den homologen Carbonylen [Fe3(CO)12] und [Os3(CO)12]. Es war gezeigt worden, das bei einem Element der ersten Übergangsreihe die d-Orbitale wenig ausladend sind. Die Überlappung mit einem Brücken-CO-Ligand ist daher oft günstiger als die direkte Metall-Metall-Bindung. Die Protonierung des Ferrats(2−) vermindert den anionischen Charakter des Clusters und führt zur Kontraktion der Metallorbitale. Als Folge wird eine der geschwächten Eisen-Eisen-Bindungen aufgegeben. Hierdurch fehlen dem Cluster nun 2 Elektronen zur 18-Elektronen-Bilanz. Diese werden nun durch den μ3-Carbonyl-Liganden ausgeglichen, indem sich dieser einem seiner drei Bindungspartner zuneigt, so dass der oben beschriebene η2-Mehrelektronen-Bindungsmodus eingenommen wird (in neuerer Nomenklatur wird aus einem CO-κC-Ligand ein CO-κC,O-Ligand).
Die zweite Protonierung erfolgt nun am O-Atom des verbrückenden Carbonyl-Liganden, der nun hinsichtlich Basenstärke und -härte der geeignete Bindungspartner für das Proton darstellt. Das dritte und vierte Proton erzeugt die Abgangsgruppe H3O+, welche sich vom Cluster löst; dieser Schritt gelingt nur unter reduzierenden Bedingungen. Hierbei wird nicht nur die Ladungsdichte erhöht – also Basizität „nachgeladen“ –, sondern der entstehende Carbidocluster genügt erst nach Aufnahme zweier Elektronen der 18-Elektronen-Regel (man beachte, dass eine plausible Elektronenzahl für den η2,μ4-COH-Ligand im Edukt nicht ohne weiteres angegeben werden kann, dass aber der entstehende Carbido-Cluster wieder eindeutig abgezählt werden kann: 4 × 8 e für 4 Fe + 12 × 2 e von 12 CO + 1 e von H + 1 e Anionladung + 4 e von C + 5 × 2 e aus 5 Fe–Fe-Bindungen = 72 e; dividiert durch 4 Fe ergibt 18 e pro Fe).
Mit dem letzten Schritt ist die CO-Aktivierung abgeschlossen, die C-O-Bindung ist vollständig aufgehoben, es ist ein Carbido-Ligand entstanden. Das nächste Proton erzeugt aus diesem einen Methylidin-Liganden. Die letzten beiden Protonen führen schließlich zur Destabilisierung und zum Zerfall des Clusters. Aus dem Methylidin-Ligand ist zum Schluss Methan geworden. (Im folgenden Schema steht das Symbol Fe im Cluster für ein Fe(CO)3-Fragment; hier das Schema als pdf.)
Bei den beiden letzten Beispielen wurde eine vereinfachte Methode verwendet, um zu prüfen, ob die 18-Elektronen-Regel erfüllt ist. Die Vereinfachung bestand darin, dass auf die Zuweisung formaler Oxidationstufen verzichtet wurde. Das Verfahren ist überall dort üblich, wo die Oxidationsstufe des Zentralmetalls keine besondere Bedeutung hat wie in weiten Bereichen der Organometallchemie. Solange nur die Frage nach der Gesamtelektronenbilanz eines Metallkomplexes interessiert, ist es belanglos, welches Zählverfahren angewendet wird. Wichtig ist nur, die Regeln der verschiedenen Verfahren nicht zu vermischen. Zur Illustration werden die beiden oben benutzten Rechenmethoden an einigen Beispielen gegenübergestellt.
Zum Vergleich noch einmal die beiden Rezepte:
Mit formalen Oxidationsstufen: (1) Jedem Ligand wird seine von der IUPAC festgelegte Ladung zugewiesen; (2) die Oxidationsstufe des Metalls wird bestimmt; (3) die Elektronenzahlen von Zentralmetall und Liganden werden addiert. In diese Rechnung gehen geläufige Neutralmoleküle wie Amine, Phosphane, Wasser, CO, Alkene, etc. als 2e-Donoren ein; mehrzähnige Liganden werden sinngemäß behandelt. NO ist ein neutraler 3e-Donor. Anionen wie Hydrid, Halogenid, Chalkogenid, Amid, Phosphanid, Alkyl, Alkylen, Alkyliden, etc. tragen 2 Elektronen je Zentralmetall bei, maximal so viel wie sie freie Elektronenpaare in geeigneter räumlicher Ausrichtung aufweisen; π-Hinbindungen werden üblicherweise nicht mitgezählt. Ein Oxido-Ligand ist daher 2e-Donor im terminalen Bindungsmodus, 4e-Donor als μ2-Ligand und 6e-Donor als μ3-Ligand; der Hydrido-Ligand ist 2e-Donor in allen Bindungsmodi. Ein η5-Cyclopentadienyl-Ligand ist ein anionischer 6e-Donor, η6-Benzol ist ein neutraler 6e-Donor.
Die bei diesem Verfahren erhaltenen Oxidationsstufen sind formal und stimmen vor allem bei radikalischen Liganden oft nicht mit der spektroskopischen Oxidationsstufe überein. Die Bestimung der formalen Oxidationsstufe kann hier in die Irre führen. Dicarbonyl-dinitrosyl-eisen, [Fe(CO)2(NO)2], wäre demnach ein Eisen(0)-Komplex, die Elektronenkonfiguration wäre d8. Man könnte bei dieser Konfiguration und wegen der Starkfeldliganden auf die Idee kommen, der Komplex sei quadratisch-planar, er ist jedoch verzerrt tetraedrisch, wie es die spektroskopische Oxidationsstufe auch nahelegt. Um diese zu ermitteln, muss der elektronische Zustand des Liganden bekannt sein. [Fe(CO)2(NO)2] enthält nach Struktur- und spektroskopischen Daten den 2e-Donor NO+. Die spektroskopische Oxidationsstufe des Eisens ist daher −II, es ergibt sich also ein geschlossenschaliger d10-Komplex. Dass die Idee vom quadratisch-planaren Komplex nicht gut ist, zeigt sich auch bei formaler Zählweise bei der Elektronenbilanz, die unabhängig von der Verteilung zwischen Metall und Ligand ist. Mit der formalen Oxidationsstufe ergibt sich als Summe der Beiträge von Zentralmetall, den Carbonyl- und den Nitrosyl-Liganden 8 + 2 × 2 + 2 × 3 = 18. Quadratisch-planare d8-Komplexe wie [Ni(CN)4]2− weisen jedoch 16 Elektronen auf!
Die bei Clustern und in der Organometallchemie übliche Zählweise ist schneller (wenn man die etwas seltsamen Regeln verinnerlicht hat), ergibt jedoch nicht die Oxidationsstufe des Zentralmetalls – die hier jedoch meist auch wenig aussagekräftig ist (Cluster) oder nur wenig interessiert (Organometallchemie). Alle Bausteine werden als neutral angenommen. Wasserstoff, Halogenatome, Alkylreste und andere einbindige funktionelle Gruppen tragen nun 1 Elektron bei. Hinzu kommen jeweils 2 Elektronen für jede zusätzliche koordinative Bindung. Im μ2-Modus sind Halogene daher 3e-Donoren (1 normale + 1 koordinative Bindung). Ein η5-Cyclopentadienyl-Ligand ist ein neutraler 5e-Donor (1 normale Bindung + 2 koordinative Bindungen), η6-Benzol ist ein neutraler 6e-Donor (0 normale + 3 koordinative Bindungen). Ein terminales O-Atom trägt 2 Elektronen bei (1 normale Doppelbindung + 0 koordinative Bindungen), ein μ2-Oxido-Ligand in der Regel 2 (2 normale Bindungen + 0 koordinative Bindung), ein μ3-Oxido-Ligand 4 (2 normale + 1 koordinative Bindungen). Da Neutralmoleküle keine freie Valenzen für normale Bindungen aufweisen, sondern nur koordinative Bindungen ausbilden (siehe oben bei Benzol), werden sie genauso behandelt wie bei der ersten Zählmethode.
Beispiele für diese Zählweise sind bei den oben genannten Clustern gegeben, bei denen eine die Oxidationsstufen einschließende Rechnung sehr umständlich wäre. Beide Zählweisen führen zu derselben Elektronenbilanz, wie das Beispiel des Tris(μ-bromido)-hexacarbonyl-dimanganat(1−)-Ions, [(CO)3Mn(μ-Br)3Mn(CO)3]−, zeigt. Bei einer Rechnung mit Oxidationsstufen ergibt sich aus der Gesamtladung und der Ladung der mono-anionischen Bromido-Liganden +I als formale Oxidationsstufe des Metalls:
| mit Oxidationsstufe | neutrale Bausteine | |||||||
|---|---|---|---|---|---|---|---|---|
| 2 MnI | 2 × 6 | = | 12 | 2 Mn | 2 × 7 | = | 14 | |
| 6 CO | 6 × 2 | = | 12 | 6 CO | 6 × 2 | = | 12 | |
| 3 μ-Br− | 3 × 4 | = | 12 | 3 μ-Br | 3 × 3 | = | 9 | |
| [ ]− | = | 1 | ||||||
| Σ | = | 36 | Σ | = | 36 | |||
| pro Mn | : 2 | = | 18 | pro Mn | : 2 | = | 18 | |
Beide Zählweisen ergeben übereinstimmend den 18-Elektronen-Fall. Es sind also keine Mangan-Mangan-Bindungen zu formulieren. Ein Nachtrag zur Nomenklatur: Die IUPAC lässt zu, entweder die Gesamtladung (arabische Zahl + Vorzeichen) oder die formale Oxidationsstufe des Metalls (Vorzeichen + römische Zahl) zur eindeutigen Bezeichnung zu verwenden. Das Dimanganat-Ion kann also auch Tris(μ-bromido)-hexacarbonyl-dimanganat(I) genannt werden.
Die etwas gewöhnungsbedürftige Zählweise, dass gleichartige Bindungen willkürlich in kovalente und koordinative Bindungen unterteilt werden, dass also die 3 Elektronen eines μ2-Halogeno-Liganden die Summe aus 1 Valenzelektron für die Bindung eines Halogenatoms an das erste Metallatom und eines Elektronenpaars für eine koordinative Bindung zum zweiten Metallatom darstellen, spielt auch bei der Interpretation von Bindungsmodi in Clustern eine Rolle. Einer der vierkernigen Eisencluster im oben behandelten Schema ist der elektroneutrale Cluster [Fe4(μ-H)(CO)12(η2,μ-CH)]. Dessen Elektronenbilanz stimmt mit der 18-Elektronen-Regel überein, wenn wie folgt gerechnet wird:
| [Fe4(μ-H)(CO)12(η2,μ-CH)] | |||
|---|---|---|---|
| 4 Fe | 4 × 8 | = | 32 |
| 12 CO | 12 × 2 | = | 24 |
| 1 H | = | 1 | |
| 1 CH | = | 5 | |
| 5 Fe–Fe | 5 × 2 | = | 10 |
| Σ | = | 72 | |
| pro Fe | : 4 | = | 18 |
Die Behandlung des Methylidin-Liganden als 5-Elektronen-Donor geht auf das folgende Bild zurück:
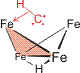
Würde das CH-Fragment nur mit seinen drei Valenzelelektronen zum Clusteraufbau beitragen, wäre die 18-Elektronen-Regel nicht erfüllt. Die Bedeutung der Regel zeigt sich nun an der Orientierung des CH-Liganden. Das H-Atom, das im geläufigen 3-Elektronen-Bindungsmodus des CH-Liganden von allen Metallatomen entfernt ist, neigt sich einem Fe-Atom zu. Hierdurch gehen die beiden Elektronen der C–H-Bindung eine koordinative Bindung zu diesem Fe-Atom ein, der CH-Ligand wird zum 3+2=5e-Donor und der Cluster wird elektronenpräzise im Sinne der Regel. Die hier dargestellte Wechselwirkung eines C–H-Elektronenpaares mit einem Metallatom wird agostische Bindung genannt.
Das Beispiel unterstreicht, dass in der Clusterchemie die Verbindungsstriche zwischen Atomen nicht notwendigerweise 2e,2z-Bindungen darstellen. Auch wenn unter Berücksichtigung von Elektronegativitäten und Oxidationsstufen der Methylidin-Baustein realistischer als CH3−-Ligand betrachtet wird, stehen für die vier eingezeichneten Fe–C-Bindungen nur drei Elektronenpaare zur Verfügung.
Die 18-Elektronenregel (die Isolobalbeziehung, die auf dieselbe Wurzel zurückgeht, führt zum gleichen Ergebnis) ist in der Regel gut erfüllt, solange weniger als ungefähr sechs Metallatome in einem Cluster enthalten sind. Oktaedrische Metallcluster folgen diesen Regeln oft nicht. Das Ligand:Metall-Verhältnis ist hier so klein, dass es sich um Elektronenmangelverbindungen handelt, die den Regeln für den Aufbau anderer Elektronenmangelverbindungen wie den Boranen folgen. Diese Wadeschen Regeln verstehen einen Cluster als Derivat eines Deltaeders, dies ist ein Polyeder, der nur von Dreiecksflächen begrenzt ist. Die trigonale Bipyramide, Oktaeder und Ikosaeder sind Deltaeder, Würfel nicht. Ein Cluster, dessen Gerüstatome ein Deltaeder bilden, ist ein closo-Cluster. Dieser tritt bei Nebengruppenmetallverbindungen auf, wenn bei n Übergangsmetallatomen 14 n + 2 Elektronen eingebracht werden. Wie bei den Wadeschen Regeln Elektronen zu zählen sind, zeigt ein Beispiel. Unter den vielen Osmiumcarbonylen und Carbonylosmaten finden sich das trigonal-bipyramidale [Os5(CO)16] und das oktaedrische [Os6(CO)18]2−. Die Elektronenzahl ergibt sich als Summe der tatsächlich vorhandenen Elektronen, das heißt, Metall-Metall-Bindungen werden nun nicht mehr explizit gezählt, da sie in den Regeln berücksichtigt sind. Das fünfkernige Carbonyl weist daher 5 × 8 + 16 × 2 = 72 Elektronen auf. Mit n = 5 ergibt sich: 14 n + 2 = 14 × 5 + 2 = 72 Elektronen. Für den Cluster wird also der closo-Typ erwartet, was auch zutrifft. Der closo-Fall sollte bei einem sechskernigen Cluster bei 14 n + 2 = 14 × 6 + 2 = 86 Elektronen auftreten. Man vergleiche mit dem Hexaosmat: 6 × 8 + 18 × 2 + 2 (die Anionladung) = 86 Elektronen.
Das oktaedrische Anion lässt sich mit der 18-Elektronenregel nicht zutreffend beschreiben. Zu der Summe von 86 Elektronen würden noch 12 × 2 = 24 Elektronen hinzugezählt, um die 12 Metall-Metall-Bindungen zu berücksichtigen. Als Summe ergibt sich 110 Elektronen; dividiert durch 6 wird 18 Rest 2 erhalten, der Cluster wäre nicht elektronenpräzise. Wie oben angemerkt, tritt bei dem nicht-oktaedrischen [Os5(CO)16] noch keine Schwierigkeit auf. Zu den 72 Elektronen der oben angestellten Rechnung wären 9 × 2 = 18 Elektronen für 9 Metall-Metall-Bindungen zu addieren. Es ergeben sich dann für das einzelne Os-Atom 90 : 5 = 18 Elektronen.
Sind mehr Elektronen vorhanden als es dem closo-Fall entspricht, entstehen offenere Strukturen, die sich von Deltaedern durch Weglassen einer oder mehrerer Ecken ableiten. Fehlt 1 Ecke, liegt eine nido-Struktur vor, 2 fehlende Ecken entsprechen einer arachno-Struktur. Die zugehörigen Elektronenzahlen sind 14 n + 4 und 14 n + 6. Kleinere Cluster folgen oft sowohl der 18-Elektronenregel als auch den Wadeschen Regeln. Ein Beispiel sind die oben behandelten Eisencluster [Fe5C(CO)15] und [Fe4(CO)13]2−, für die schon gezeigt wurde, dass sie der 18-Elektronenregel genügen.
Der Fe5-Cluster weist 5 × 8 + 15 × 2 + 4 = 74 Elektronen auf. Mit n = 5 entspricht dies dem 14n+4-Fall, es ist also eine nido-Struktur zu erwarten. Bei n = 5 wäre also nach dem Deltaeder mit n = 6 zu fragen, dem dann 1 Ecke fehlen sollte. Dies trifft zu: das Fe5-Gerüst hat die Struktur eines Oktaeders mit einer fehlenden Ecke. Analog stellen die 4 × 8 + 13 × 2 + 2 = 60 Elektronen des Tetraferrats die 14n+4-Bilanz des nido-Falls für n = 4 dar. Entsprechend fehlt dem einfachsten Deltaeder, der trigonalen Bipyramide, 1 Ecke, wodurch sie zum Tetraeder wird.
Eine arachno-Struktur ist bei [Fe4(μ-H)C(CO)12]− zu erwarten. Dieser der 18-Elektroneregel entsprechende Cluster enthält 4 × 8 + 12 × 2 + 1 (H) + 4 (C) +1 (Ladung) = 62 Elektronen. Mit n = 4 ist dies der 14n+6-Fall. Vom Deltaeder mit n = 6 wären also 2 Ecken abzutrennen. Auch dies trifft zu. Die Struktur des Fe4-Gerüsts des Carbido-Clusters wird erhalten, wenn von einem Oktader 2 benachbarte Ecken entfernt werden.
16e-d8-Komplexe bilden neben großen Clustern eine wichtige Ausnahme von der 18e-Regel. Im Gegensatz zum stark antibindenden x2−y2-Orbital, das unbesetzt bleibt, ist das z2-Orbital durch das Fehlen der beiden Liganden, mit denen im Oktaeder eine antibindende Wechselwirkung vorläge, nicht destabilisiert. Durch die Besetzung des z2-Orbitals mit zwei Elektronen enthält ein quadratisch-planarer d8-Komplex zwei metallständige Valenzelektronen mehr als ein oktaedrischer Komplex. Das gefüllte, in den Raum hinausragende z2-Orbital gibt dem Metallzentrum Basizität, außerdem ist es Angriffspunkt für Oxidationsreaktionen. Solche Reaktionen sind in großem Umfang mit „Vaskas Komplex“, trans-Bis(triphenylphosphan)-carbonyl-chlorido-iridium(I), trans-[Ir(PPh3)2(CO)Cl)], untersucht worden. Die Metallbasizität zeigt sich zum Beispiel bei der Reaktion mit NO+BF4−, bei dem ein Lewis-Säure/Base-Addukt entsteht, in dem das Iridium-Zentrum als Lewis-Base eintritt:
Man beachte, dass bei üblicher Zählweise das bindende Elektronenpaar zwischen Ir und N dem elektronegativeren Stickstoff zuzuweisen wäre, dass also ein NO−-Ligand vorliegt – NO+ ist oxidativ addiert worden. Die Anbindung ist nicht linear wie in dem besonderen Fall des [Fe(H2O)NO]2+-Ions, bei dem ein Triplett-NO− diskutiert wurde. Die hier vorliegende gewinkelte Ir-N-O-Gruppe (124° im Tetrafluoridoborat des abgebildeten Ions; in dessen Kristallstruktur liegt das Anion in räumlicher Nähe zum Komplexkation vor, so wie dies im Schema angedeutet ist) entspricht vielmehr dem häufiger beobachteten Fall eines Singulett-NO−-Liganden, der durch eine Lewis-Formel der Art
angemessen beschrieben wird (die letzten drei Formeln als pdf). Ist das Gegenion des Elektrophils ein besserer Ligand als das Tetrafluoridoborat-Ion, so ergänzt dieses die Koordinationsspäre des Iridiums. Dies gilt auch für die Abgangsgruppe bei der Reaktion von Vaskas Komplex mit polaren Substraten wie den Halogenwasserstoffen oder Iodmethan (im folgenden Schema rechts; pdf):
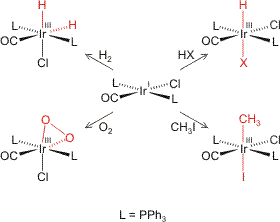
Die mit HX oder CH3I ablaufende oxidative Addition an ein polares Substrat wird durch einen nucleophilen Angriff des basischen Metallzentrums auf den positiv polarisierten Molekülteil des Substrats eingeleitet. Bei Iodmethan entspricht dies der bekannten SN2-Reaktion mit einem Nucleophil, bei der Iodid als Abgangsgruppe fungiert. Bei der Umsetzung mit Vaskas Komplex füllt das Iodid-Ion die Koordinationslücke am formalen Iridium(III)-Zentrum auf, und zwar in trans-Position zum eingetretenen Elektrophil (das kinetische trans-Produkt lagert sich bisweilen zum cis-Produkt um, wenn dieses stabiler ist). Man beachte die Ursache der 2-Elektronen-Oxidation: das Elektrophil bindet in der Regel über ein Atom an das Metallzentrum, das elektronegativer als das Zentralmetall ist. Das vom basischen Metallzentrum eingebrachte Elektronenpaar wird im entstehenden Lewis-Säure-Base-Addukt dem elektronegativeren Ligand zugerechnet – das Metall hat wie im Fall der NO+-Addition formal 2 Elektronen abgegeben.
Auf der linken Seite des Schemas sind zwei Reaktionen angeführt, die ebenfalls einen wichtigen Elementarschritt der Organometallchemie darstellen, die oxidative Addition an ein unpolares Substrat. Diese Reaktion wird oft einfacher als cis-Addition bezeichnet. Die Addition verläuft hier prinzipiell verschieden. Das unpolare Substrat nähert sich hier mit seiner Element-Element-Bindung dem Metallzentrum. Anschließend bilden sich in einer konzertierten Reaktion die beiden neuen Metall-Element-Bindungen. 2 Elektronen der neuen Bindungen stammen vom bindenden Elektronenpaar des Substrats, 2 Elektronen stammen vom Metall. Auch hier handelt es sich um eine 2-Elektronen-Oxidation, da alle 4 bindenden Elektronen der beiden Metall-Element-Bindungen nach der Reaktion den elektronegativeren Nichtmetall-Atomen zugewiesen werden. Die Umkehrreaktion der cis-Addition ist die reduktive Eliminierung.
Die cis-Addition verläuft nicht nur mechanistisch anders als die Addition eines polaren Substrats, auch die elektronischen Voraussetzungen unterscheiden sich. 18-Elektronen-Komplexe sind zur cis-Addition nicht in der Lage – sie hätten nach der Reaktion 20 Elektronen. Der elektrophile Teil polarer Substrate kann jedoch oxidativ addiert werden, auch ohne dass anschließend eine Abgangsgruppe koordiniert. Beispiele wurden schon behandelt. So ist neben der NO+-Anlagerung die Protonierung von Carbonylmetallaten eine oxidative Addition ohne Veränderung der gesamten Elektronenbilanz. Man überzeuge sich anhand der Reaktionen
[Fe(CO)4]2− + H+ = [HFe(CO)4]− und
[HFe(CO)4]− + H+ = [H2Fe(CO)4]
Die in Katalysecyclen wichtige cis-Addition ist demnach nur zu erwarten, wenn eine um zwei Einheiten höhere Oxidationsstufe zur Verfügung steht und zugleich in der oxidierten Form zwei Koordinationsstellen frei sind. Diese Bedingungen werden vor allem von quadratisch-planaren d8-Komplexen erfüllt.
Es ist nicht trivial, deren Auftreten abzuschätzen. d8-Zentren, die ein 4d- oder 5d-Metall, und neben Donor/Acceptor- auch noch Halogeno-Liganden enthalten, sind zuverlässig quadratisch-planar. Neben diesen Lehrbuchbeispielen gibt es vor allem bei den in Katalysecyclen wichtigen reaktiven Zwischenstufen einige Unsicherheit.
Ein Beispiel ist [HCo(CO)3], das im Katalysecyclus der Oxo-Synthese auftritt und das durch CO-Abspaltung aus dem 18-e-Komplex [HCo(CO)4] entsteht. Die Struktur von [HCo(CO)3] ist in der Literatur umstritten. In einer neueren Arbeit (C.-F. Huo, Y.-W. Li, G.-S. Wu, M. Beller, H. Jiao, J. Phys. Chem. A 2002, 106, 12161–12169) ergibt sich als stabilste Form ein planares Molekül im Singulett-Zustand. Dessen C2v-Symmetrie ergibt sich aus einer quadratisch-planaren Anordnung, bei der die zum H-Atom cis-ständigen CO-Gruppen im Sinne des VSEPR-Modells etwas vom trans-CO weg und damit zum H-Atom hingebogen sind. Die hier gezeigten Regeln (CoI als d8-Zentrum, 4 Liganden, 16e insgesamt → quadratisch-planare Anordnung) führen also zum richtigen Ergebnis – falls nicht bei weiteren Rechnungen wieder alles anders wird.
Dieses Ergebnis ist jedoch eher unerwartet. So ist das verwandte, bei der Photolyse von [Fe(CO)5] entstehende [Fe(CO)4] weder dem experimentellen Befund noch der Rechnung nach quadratisch-planar [16e1]. Die Struktur des Singulett-Zustands, dem diese Geometrie zugetraut werden kann, ist vielmehr eine trigonale Bipyramide, der einer der drei äquatorialen Liganden fehlt. Überdies ist das Singulett nicht der Grundzustand sondern ein Triplett mit stark verzerrter tetraedrischer Geometrie – ganz im Sinne des Isolobalkonzepts, bei dem Sie 16e-Metallcarbonyle als zweibindinge Fragmente kennenlernen. Für den Fortgang von Reaktionen sind solche Ergebnisse von Bedeutung. Für ein [Fe(CO)4] im Singulettzustand ist die oxidative Addition von H2 zum bekannten [H2Fe(CO)4] ohne weiteres zu erwarten, für ein Triplett-[Fe(CO)4] ist die Reaktion jedoch spinverboten [16e2].
J. N. Harvey, R. Poli: Computational study of the spin-forbidden H2 oxidative addition to 16-electron Fe(0) complexes. Dalton Trans. 2003, 4100–4106 [16e2].
E. Hunstock, C. Mealli, M. J. Calhorda, J. Reinhold: Molecular Structures of M2(CO)9 and M3(CO)12 (M ) Fe, Ru, Os): New Theoretical Insights. Inorg. Chem. 1999, 38, 5053–5060 [cluster1].