 Das Liebig-Laboratorium
Das Liebig-Laboratorium
Organisation
Das Liebig-Laboratorium ist ein gemeinsames Praktikum der Lehrbereiche Anorganische Chemie und Physikalische Chemie für das 1. Fachsemester im Bachelorstudiengang Chemie und Biochemie. Die Zuständigkeiten finden Sie bei der Projektübersicht. Die Laborräume befinden sich im 1. Stockwerk von Haus D.
Das Praktikum beginnt am 31. Oktober 2022 und findet montags, mittwochs und donnerstags von 13:00–17:00 Uhr sowie freitags von 12:30–15:30 Uhr statt. Alles, was an einem Tag gemacht wurde, bleibt am Platz stehen. Der Gruppenassistent geht ca. 1 Stunde vor Praktikumsende herum und prüft stichprobenartig, ob das praktisch Ausgeführte verstanden wurde (zum Beispiel Reaktionsgleichungen aufstellen lassen). Ca. eine halbe Stunde vor Praktikumsende gibt es eine Schlussbesprechung, in der wiederkehrende Schwierigkeiten besprochen werden.
Begleitende Vorlesung und Literatur
Vorlesungen
Der Stoff der drei Projekte aus der Anorganischen Chemie und der Elektrochemie wird in der Vorlesung von Prof. Böttcher behandelt.
Lehrvideos und den Stoff zu den Projekten „Farben“ und „Reaktionskinetik“ finden Sie auf der Moodle Seite der Vorlesung "Einführung in die experimentelle Chemie". Der Einschreibeschlüssel wird in der Vorlesung bekannt gegeben.
Literatur
Abkürzungen im Text im Stil Mor-489K28.9 beziehen sich auf Mortimers Chemie. Lesen Sie die Abkürzung so: „Mortimer, Seite 486, Kapitel 28.9“. Die Verweise beziehen sich auf die 10. Auflage:
C. E. Mortimer, U. Müller: Chemie. 10. Auflage, Thieme 2010 (ISBN 978-3-13-484310-1), die Sie innerhalb des Münchner Hochschulnetzes auch als „E-Book“ lesen können.
Physikalisch-chemische Inhalte finden Sie im „Atkins“:
P. W. Atkins, J. de Paula: Physikalische Chemie. 4. Auflage, Wiley-VCH 2006.
Laborsicherheit
Machen Sie sich noch mal mit allen Aspekten der Laborsicherheit vertraut, die im Skript zum Vorkurs zusammengestellt sind.
Aufbau der Projekte
Zum Kennenlernen der Projekte dienen Vorversuche, bei denen kurz Ausführung und Beobachtung protokolliert werden, außerdem wird eine Reaktionsgleichung formuliert.
Oft schließen sich dann Übungsanalysen an, die zum Kennenlernen der Methoden einer nachfolgenden Vollanalyse dienen.
Vollanalysen betreffen meist Proben aus der realen Umwelt. Die Bewertung ist ähnlich, nur dass Sie bei einer Vollanalyse im Anschluss an die Eingabe Ihres Ergebnisses den Sollwert durch eine automatische Analysemethode selbst bestimmen.
Das Ergebnis wird in einem kurzen gemeinsamen Protokoll zusammengestellt und es wird kurz darüber berichtet. Am Schluss des Protokolls wird zusammengestellt, wie die erwähnten Stoffe und Phänomene auf Englisch heißen.
Projektübersicht
Die ersten drei Projekte werden vom Lehrbereich Anorganische Chemie (Prof. Ivanovic-Burmazovic, Prof. Böttcher) betreut.
Die letzten drei Projekte werden vom Lehrbereich Physikalische Chemie (Prof. Müller-Caspary) betreut.
Die zeitliche Abfolge der einzelnen Projekte während des Praktikums wird individuell geregelt, um die zur Verfügung stehenden Geräte optimal zu nutzen. Im WS 2022/2023 werden in allen Sälen ab dem 31. Oktober Kapitel 1–3 bearbeitet, ab dem 28. November bearbeiten alle Säle die Kapitel 4–6.
Der Kalkkreislauf
Mit der Kohlensäure wird eine mehrbasige schwache Säure eingeführt. Kalk und Calciumhydrogencarbonat eignen sich zu Betrachtungen rund um die Begriffe Löslichkeit, Löslichkeitsprodukt und pH-Abhängigkeit der Löslichkeit. Die Bestimmung der „Wasserhärte“ führt zur Komplexometrie.
Säure und Base und Chelatligand: Aminosäuren als polyfunktionelle Moleküle
Obwohl sie recht kleine Moleküle sind, enthalten Aminosäuren zahlreiche unterschiedliche funktionelle Gruppen. Im Mittelpunkt des Projekts steht das Zusammenspiel saurer und basischer Funktionen, durch das Aminosäuren zu Ampholyten werden. Deren typische Titrationskurven setzen sich aus zwei Einzelkurven zusammen. Die Bildung stabiler Chelatkomplexe lernen Sie als Konkurrenz zwischen Protonen und Metallionen um die Bindungsstellen am Aminosäuremoleküle kennen. Ein Aminosäure-Anion mit sechs funktionellen Gruppen bindet Metalle besonders fest: edta.
Bleiche, Desinfektion, oxidativer Stress: starke Oxidationsmittel
Die vielfältige und praktisch bedeutende Redoxchemie des Wasserstoffperoxids und seiner Derivate bildet den roten Faden durch das Projekt, bei dem Grundlegendes rund um Redoxreaktionen wie Oxidationszahlen, Redoxgleichungen, pH-Abhängigkeit von Redoxreaktionen vermittelt wird. Als weiteres starkes Oxidationsmittel kommt Permanganat hinzu.
Farbe
Woher kommen Farbeindrücke? Farbreaktionen und -eindrücke bestimmen das tägliche Leben und sind auch ein wesentlicher Bestandteil der Analytischen Chemie. Tatsächlich haben „Farben“ ganz verschiedene Ursachen. In diesem Projekt werden grundlegende Phänomene, die zu Farbeindrücken führen, unterschieden und Messmethoden vorgestellt.
Reaktionskinetik
Die Explosion von Nitroglycerin verläuft in Sekundenbruchteilen. Die Umwandlung von Graphit in Diamant verläuft dagegen unmessbar langsam (findet jedoch statt). In diesem Projekt soll ein erster Einblick in die Welt der Reaktionskinetik gegeben werden, indem der abstrakte, mathematische Begriff der Reaktionsordnung an einfachen Beispielen veranschaulicht und greifbar wird.
Elektrochemie
Elektrochemie bezeichnet mehrere verschiedene Teilgebiete innerhalb der Chemie. Sie ist zum einen eine Synthesemethode als präparative Elektrochemie, Elektrolyse oder Elektrosynthese, zum anderen ist sie ein Teilgebiet der physikalischen Chemie, welches sich mit dem Zusammenhang zwischen elektrischen und chemischen Vorgängen befasst. Die technische Chemie kennt neben großtechnisch angewandten elektrochemischen Synthesemethoden noch die Batterie- und Brennstoffzellentechnik, sowie die Galvanotechnik.
Analysen
Analysen sind unter der Versuchsnummer mit einem Zusatz wie zum Beispiel
Analyse 3: Calcium-Bestimmung
markiert.
Durch die Bearbeitung der Analysen sollen Sie lernen, eigenständig und sauber zu arbeiten. Machen Sie sich vor der Analyse Ihrer Probe mit der jeweiligen Methode vertraut. Als Hilfe stehen in den Praktikumssälen Lösungen aus, die einen bekannten Gehalt an Analyt enthalten. Versuchen Sie, diese Gehaltsangabe durch eine eigene Bestimmung zu reproduzieren und lernen Sie so zum Beispiel den Umschlagspunkt einer Titration kennen. Bearbeiten Sie erst dann Ihre Analyse. Beachten Sie weitere Hinweise, die am Schluss der einzelnen Projekte zusammengestellt sind. Ein wichtiger Punkt: Eine Analyse kann genau einmal zur Bewertung vorgelegt werden.
Teilen Sie das Ergebnis einer Analyse über Ihr grünes Analysenheft mit. Das Ergebnis der Analysen trägt zu den Teilnoten für den Vorkurs sowie der drei Projekte aus der Anorganischen Chemie bei. In der Regel gilt: Note 1 bei einer Abweichung bis 1 mg vom Sollwert, Note 2 bei 2 mg, Note 3 bei 3 mg, Note 4 bei 4 mg, nicht bestanden ab 4,1 mg).
Forschungsprojekte im WS 2022/2023
Jede Gruppe bekommt am Donnerstag, dem 22. Dezember 2022, in ihrem Saal ein Thema zugelost und bearbeitet dieses vom 09.–13. Januar 2023 in der gewohnten Praktikumszeit. Die Fragestellungen in den Versuchsanleitungen bilden die Grundlage der Forschungsprojekte. Zusätzlich ist Ihre Eigeninitiative gefragt. Dies bedeutet, dass Sie selbst die Fragestellungen erweitern und, wenn notwendig, auch die Versuchsdurchführung entsprechend anpassen können.
Am 19. Januar 2023 findet für alle Gruppen in den Praktikumssälen eine Posterpräsentation statt. Jede Gruppe bereitet hierfür ein DIN-A0-Poster mit den Fragestellungen, den Lösungswegen und den Ergebnissen vor. Die Gestaltung der Poster ist frei. Während der Posterpräsentation stellt jede Gruppe in einem 15 Minuten dauernden Vortrag ihr Poster den anderen Gruppen im Saal vor. Dabei erfolgt die Benotung durch die Dozenten. Im Anschluss sollen Sie sich auch ein Bild von den Ergebnissen und Postern aus den anderen Sälen machen.
Benotung
Die Vorlesung zum Praktikum (3 Leistungspunkte) und das Praktikum (9 CPs) sind eigenständige Lehrveranstaltungen, deren Noten nicht zusammengefasst werden.
Die Vorlesung zum Praktikum wird von einer Klausur begleitet. (Notenschlüssel). Wird die Klausur nicht bestanden, besteht die Möglichkeit, an einer Wiederholungsklausur teilzunehmen.
Die 1. Klausur zur Praktikumsvorlesung findet am 08. Februar 2023 von 13:00–15:00 Uhr im Liebig- und Buchner-Hörsaal statt. Die Wiederholungsklausur findet am 31. März 2023, 10:00–12:00 Uhr (Liebig-Hörsaal) statt.
Beachten Sie, dass Sie erst zu den regulären Klausurterminen des darauffolgenden Wintersemesters die nächste Gelegenheit zur Klausurteilnahme haben, falls Sie auch die Wiederholungsklausur nicht bestehen.
Die Praktikumsnote setzt sich aus den folgenden Teilnoten zusammen: jeweils ein Viertel für (1) die Leistung im Vorpraktikum, (2) die drei Projekte aus der Anorganischen Chemie, (3) die drei Projekte aus der Physikalischen Chemie und (4) das Protokoll und die Vorstellung des Forschungsprojekts.
Technisches
Prüfen Sie hier, ob Ihr Browser das Skript korrekt darstellt.
Zum Drucken verwenden Sie am besten eine pdf-Version dieses Textes. Wenn Sie am Praktikum teilnehmen, erhalten Sie von uns zu Beginn des Praktikums einen Ausdruck des Skripts.
… noch ein paar Tipps, wie Sie Fehler vermeiden können:
Einen solchen Kasten finden Sie am Ende mancher Projekte. Hier haben wir typische Fehler zusammengestellt, die uns in früheren Praktika aufgefallen sind – vor allem bei der Bearbeitung der Analysen. Am Besten lesen Sie diese Kommentare, bevor Sie die praktische Arbeit am Projekt beginnen.
Kurzanleitung zur Bedienung des Titrierautomaten
Der Metrohm-Titrierautomat Titrando ermöglicht die Durchführung einer automatisierten Titration gekoppelt mit der potentiometrischen Messung von pH-Wert, Ionenkonzentration oder Redoxpotentialen. Je nachdem welche Messgröße im Experiment bestimmt werden soll, kommen dabei unterschiedliche Elektroden zum Einsatz. Der Anschluss des Titrandos an einen PC erlaubt es, mit Hilfe der Software tiamo 2.0, die Messergebnisse auszuwerten.
Die folgende Abbildung zeigt den allgemeinen Aufbau eines Titrandos.

Die in diesem Praktikum verwendete Titrando-Version besteht im wesentlichen aus einem Titrierstand für die Maßlösung (A), einer Dosiereinheit (20 mL) mit Dosierschlauch (B), einem Rührer (C), je nach Messmethode unterschiedlichen Elektroden (D) und einem PC mit der Titrationssoftware tiamo 2.0 (E).
Die nachfolgende Abbildung zeigt den Aufbau zur Durchführung der automatischen Titration. Die zu titrierende Probe wird in einem hohen 150-mL-Becherglas auf ein Gesamtvolumen von 100 mL aufgefüllt. Die Elektrode taucht mittig, bis kurz über den Rührfisch, in die Lösung ein, die Dosierspitze wird am Rand bis kurz über den Boden des Becherglases eingetaucht. Es muss darauf geachtet werden, dass der Rührfisch weder an der Elektrode noch an der Dosierspitze anschlägt. Nur mit dieser Anordnung ist eine erfolgreiche Titration möglich!

Titrationssoftware tiamo 2.0
Die zur Steuerung des Titrando sowie zur Auswertung der Messergebnisse benötigte Software tiamo 2.0 kann mit einem Doppelklick auf die entsprechende Verknüpfung auf dem Desktop geöffnet werden. Während des Praktikums sind für Sie drei Oberflächen von Bedeutung; der Arbeitsplatz, die Datenbank und die Manuelle Bedienung. Alle drei Oberflächen lassen sich über die jeweiligen Schaltflächen am linken Bildrand erreichen.
Im Arbeitsplatz geben Sie ihre jeweiligen Probedaten an, starten die Titration und verfolgen ihren Ablauf in Echtzeit. In der Datenbank werden die Messwerte der durchgeführten Titrationen gespeichert und können jederzeit abgerufen werden. Die manuelle Bedienung dient unter anderem zum Befüllen und Leeren der Dosiereinheit. Dies ist vor allem beim Wechsel der Maßlösung von Bedeutung.
Anschließen oder Wechseln der Maßlösungen
Um die gewünschte Maßlösung anzuschließen, entleeren Sie zunächst die in der Dosiereinheit vorliegende Flüssigkeit. Öffnen Sie hierzu die Oberfläche „Manuelle Bedienung“ über die Schaltfläche am linken Bildrand.
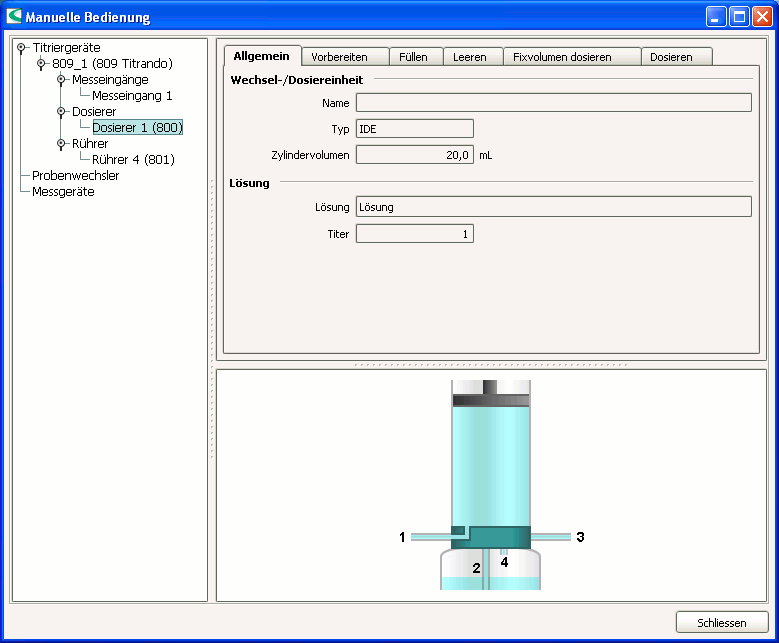
Wählen Sie im linken Unterfenster „Dosierer 1“ und anschließend im rechten Bedienfeld die Registerkarte „Leeren“ und drücken Sie auf „Start“. Spülen Sie die Dosiereinheit nun zunächst mit destilliertem Wasser. Schrauben Sie dazu das Gefäß mit der momentan angeschlossenen Maßlösung ab und ersetzen Sie diese durch destilliertes Wasser. Wählen Sie die Registerkarte „Vorbereiten“ und drücken „Start“. Sobald der Vorgang abgeschlossen ist, wählen Sie die Registerkarte „Fixvolumen dosieren“. Wählen Sie zum Spülen ein Volumen von 20 mL aus und drücken Sie wieder auf „Start“. Entleeren Sie die Dosiereinheit erneut, wechseln das destillierte Wasser gegen die gewünschte Maßlösung und bereiten die Dosiereinheit wie oben beschrieben vor.
Um Verschmutzungen und korrosive Einflüsse auf die Dosiereinheit zu vermeiden, spülen Sie diese mit destilliertem Wasser, falls der Titrando für längere Zeit nicht benutzt werden sollte.
Anschließen oder Wechseln der Elektroden
Zum Anschließen einer bestimmten Messelektrode schrauben Sie zunächst den Deckel am oberen Ende der Elektrode ab und bringen statt dessen den Steckkopf des Elektrodenkabels an. Das Elektrodenkabel wird an der Rückseite des Titrandos an der Buchse „Ind.“ angeschlossen.
Neben den Messelektroden ist am Titrando noch eine Silber/Silberchlorid-Bezugselektrode angeschlossen (Buchse „Ref.“). Diese kann auch für den Fall, dass sie nicht benötigt wird, angesteckt bleiben.
Durchführung einer Titration
Überprüfen Sie zunächst, ob die entsprechende Maßlösung und die benötigten Elektroden am Gerät angeschlossen sind. Öffnen Sie dann in der Titrationssoftware die Oberfläche „Arbeitsplatz“. Wählen Sie dort im Fenster „Ablauf“ unter „Einzelbestimmung“ zunächst die gewünschte Methode aus und geben dann die weiteren Probedaten ein.
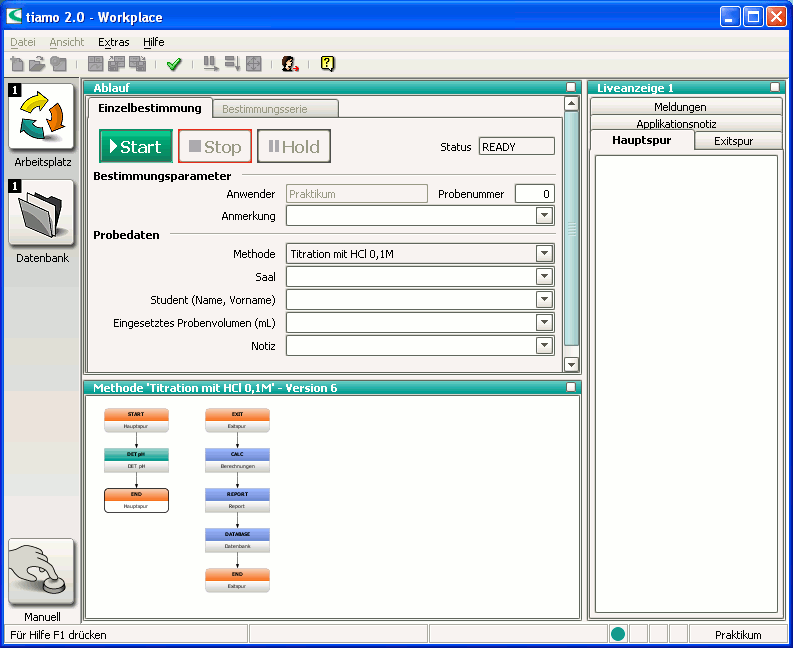
Bereiten Sie nun Ihre Probelösung entsprechend der Vorschrift aus dem Praktikumsskript vor. Einzelheiten zur Darstellung der Probelösung und eventuell verwendeter Hilfslösungen finden Sie auch rechts im Fenster „Liveanzeige“ unter der Registerkarte „Applikationsnotiz“. Stellen Sie nun die Probelösung auf den Rührer, tauchen Sie Elektrode(n) und Dosierschlauch in die Lösung und drücken Sie im Fenster „Ablauf“ auf „Start“. Unter der Registerkarte „Hauptspur“ im Fenster „Liveanzeige“ können Sie den Verlauf der Titration verfolgen. Nach Abschluss der Titration werden die Messwerte in einer Datenbank gespeichert. Um die Ergebnisse aufzurufen, öffnen Sie die Oberfläche „Datenbank“ über die entsprechende Schaltfläche am linken Bildschirmrand.
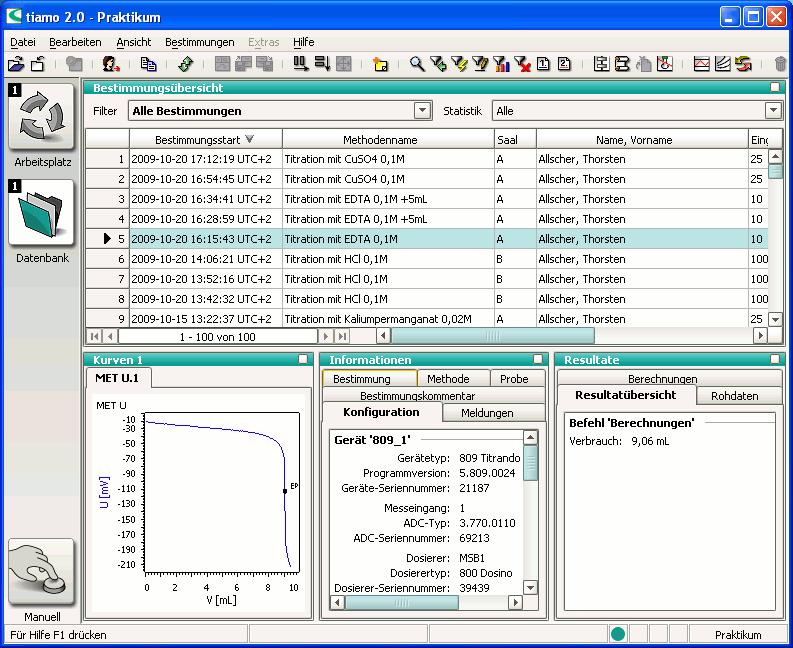
Im oberen Fenster können Sie die gewünschte Bestimmung aufrufen, die unteren Fenster zeigen dann die dazugehörige Titrationskurve, allgemeine Informationen zur durchgeführten Titration, sowie die zusammengefassten Resultate.
Messelektroden
Je nachdem, welcher Messwert bei der durchgeführten potentiometrischen Titration (Potentiometrie) erfasst werden soll, werden unterschiedliche Elektroden verwendet. Die Messanordnung besteht dabei immer aus einer Messelektrode (Indikatorelektrode) und einer Bezugselektrode (Referenzelektrode). Die beiden Elektroden werden als Halbzellen bezeichnet, welche zusammen in Lösung ein bestimmtes (aus mehreren Einzelpotentialen bestehendes) Potential liefern. Aus diesem Potential lassen sich dann pH-Wert, Redoxpotentiale oder auch Ionenkonzentrationen berechnen.

Elektroden zum Titrando. Von links: Cu-ISE, Ag/AgCl/KCl-Bezugselektrode, pH-Glaselektrode, Pt-Elektrode.
Die Kupferelektrode (Cu-ISE)
Die Kupferelektrode ist eine sogenannte ionenselektive Elektrode (ISE), die auf ein bestimmtes Ion in einer Lösung anspricht, bei der Cu-ISE Cu2+-Ionen. Die Elektrode besitzt dazu eine Kristallmembran (Kupferkristall) am unteren Ende der Elektrode. Dieser Kupferkristall sollte nicht angefasst oder dem direkten Sonnenlicht ausgesetzt werden. Um den Kristall zu schützen, befindet sich am unteren Ende eine Kappe über der Elektrode, die jedoch vor der Titration entfernt werden muss.
Kupferionen in der unter zu untersuchenden Lösung können in die Membran eindringen und so eine Potentialänderung bewirken. Als Bezugselektrode wird eine Silber/Silberchlorid-Elektrode (Ag/AgCl/KCl) verwendet, die mit einer 3 m Kaliumchlorid-Lösung gefüllt ist. Entfernen Sie während der Verwendung der Bezugselektrode den kleinen Gummistopfen am oberen Ende der Elektrode. Zur Lagerung muss der Gummistopfen wieder eingesteckt werden und die Elektrode in die Schutzhülle mit Kaliumchlorid-Lösung getaucht werden.
Die Kupferelektrode kann für die komplexometrische Titration von Metallionen verwendet werden. Da es für die komplexometrische Titration keine Elektrode gibt, die direkt auf edta anspricht, muss der Endpunkt indirekt detektiert werden. Dazu eignet sich die Kupferelektrode, wenn man die zu untersuchenden Probelösung mit wenig Kupfer-edta-Komplex versetzt. Da der Kupfer-edta-Komplex aufgrund seiner hohen Komplexbildungskonstante stabil ist, liegt in der reinen Lösung nur ein kleiner Anteil an freien Kupferionen vor. Durch Zugabe eines weiteren Metallions wird der Anteil an freien Kupferionen erhöht, welche dann in die Kupfermembran eindringen und so eine Potentialänderung herbeiführen. Auf diese Weise können die folgenden Metallionen bestimmt werden: Al3+, Ba2+, Bi3+, Ca2+, Co2+, Fe3+, Mg2+, Ni2+, Pb2+, Sr2+ und Zn2+.
Die pH-Glaselektrode (Unitrode)
Die pH-Glaselektrode (Unitrode) eignet sich für Messungen im pH-Bereich von 0–14, in einem Temperaturbereich von 0–100 °C. Die Elektrode besitzt eine Glasmembran aus einem Silicat-Grundgerüst, welches Lithiumionen enthält. Wird die Glasoberfläche in eine wässrige Lösung getaucht, so bildet sich auf der Glasoberfläche eine dünne Quellschicht (Gelschicht) aus, in der die Struktur des Glases aufgeweicht ist. Diese Schicht hat einen Durchmesser von ca. 0,1 μm (0,0001 mm). Die Glaselektrode ist innen mit einer Pufferlösung (pH = 7) gefüllt, wodurch auf der Innenseite der Glasmembran eine konstante Protonenkonzentration vorliegt. Ändert sich die Protonenkonzentration der Probelösung, kommt es an der äußeren Quellschicht der Glasmembran zu einem Ionenaustausch, was eine Potentialänderung zur Folge hat. Bei der Glaselektrode handelt es sich um eine sogenannte kombinierte pH-Elektrode, das heißt, die Bezugselektrode ist bereits mit der Messelektrode kombiniert. Es ist daher (im Gegensatz zur Cu-ISE) nicht nötig, eine zweite Elektrode als Bezugselektrode anzuschließen. Da der pH-Wert temperaturabhängig ist, besitzt die Glaselektrode auch einen eingebauten Temperatursensor. An der Oberseite der Glaselektrode befindet sich ein kleiner Gummistopfen, welcher während der Titration entfernt wird. Zur Lagerung muss der Stopfen wieder eingesteckt und die Elektrode in die mit Aufbewahrungslösung gefüllte Schutzhülle getaucht werden, um ein Austrocknen der äußeren Quellschicht zu verhindern. Die pH-Glaselektrode eignet sich sowohl zur einfachen Bestimmung des pH-Wertes, als auch zur potentiometrischen Säure-Base-Titration.
Die Platinelektrode (kombinierte Pt-Ring-Elektrode)
Sind in der Probelösung Ionen des Metalls, aus welchem die Metallelektrode besteht (Halbzelle), so stellt sich in Abhängigkeit von der Konzentration der Ionen ein Gleichgewicht an der Metalloberfläche ein, es bildet sich eine elektrochemische Doppelschicht aus. Das für das konzentrationsabhängige Gleichgewicht charakteristische Potential ist das Halbzellpotential. Sind in der Probelösung keine Ionen des entsprechenden Metalls enthalten, so können Metallelektroden dennoch ein Potential aufbauen, sofern in der Probelösung eine Redoxreaktion stattfindet. Die Elektrodenoberfläche ist dann inert gegenüber der Redoxreaktion und dient lediglich als Katalysator. Die chemische Inertheit von Platinelektroden kann man sich daher für Messung von Redoxpotentialen zunutze machen.
Kurzanleitung zur Bedienung des UV/VIS Spektrometers
Das Ocean Optics Spektrometer ermöglicht die Messung von Absorptions- und Fluoreszenzspektren sowie die spektrale Charakterisierung unterschiedlichster Lichtquellen. Gesteuert wird das Spektrometer über ein NETBOOK (user: student; pw: liebiglab) und die Software SpectraSuite.
Aufbau der Messaparatur
Bauen Sie die Messapparatur wie in der Abbildung dargestellt auf:

Nach dem Verbinden des USB-Kabels mit dem Netbook starten Sie das Programm „SpectraSuite“ vom Desktop aus und Sie erhalten folgenden Startbildschirm:.
Beachten Sie, dass das Spektrometer bereits fortwährend Daten aufnimmt (erkennbar an der gedrückten Play-Taste rechts oben). Folgende in der Menüleiste enthaltene Buttons sind für die Messungen relevant:

- Play: Kontinuierliche Aufnahme von Spektren (
 )
)
- Pause: Stoppt die Aufnahme. Das letzte Spektrum wird angezeigt (
 )
)
- Play/Pause: Aufnahme eines einzelnen Spektrums
- Strobe/Lamp Enable: An- und Ausschalten der Lichtquelle
- Dunkelstromkorrektur: Option zur Verbesserung der Messung (immer angewählt lassen)

- Integrationszeit: Zeit in der der Detektor Photonen für ein Spektrum zählt
- Scans zur Mittelwertbildung: Anzahl der Spektren über die gemittelt wird
- Boxcar Breite: Anzahl der Pixel über die gemittelt wird
Aufnahme von Spektren
Unabhängig vom eingestellten Aufnahmemodus wird die Aufnahme der Spektren über die drei Schalter in der Menüleiste gesteuert (Play, Play/Pause, Pause). Eine sinnvolle Herangehensweise ist zunächst die kontinuierliche Aufnahme zu starten und sobald das gewünschte Ergebnis erreicht ist das Spektrum durch Drücken des Schalters Pause festzuhalten. über die verschiedenen Zoomwerkzeuge
 kann ein gewünschter Ausschnitt herausgestellt werden. Anschließend kann es, wie im Abschnitt Speichern von Spektren beschrieben, gespeichert werden.
kann ein gewünschter Ausschnitt herausgestellt werden. Anschließend kann es, wie im Abschnitt Speichern von Spektren beschrieben, gespeichert werden.
Aufnahme von Emissionsspektren

Um Emissionsspektren aufzunehmen muss die Faser wie im Bild dargestellt mit dem Probenhalter verbunden werden. Ein Neutraldichte-Filter muss nicht eingesetzt werden. Verwenden Sie für die Messung eine 4-Wege-Küvette.
Achten Sie darauf, dass die aktuelle Messung im Scope-Modus [y-Achsenwert: Intensität (Counts)] läuft. Notfalls diesen über
![]() aktivieren. Zur Aufnahme eines Emissionsspektrums eignen sich folgende Einstellungen, die während der gesamten Messreihe unverändert bleiben müssen:
aktivieren. Zur Aufnahme eines Emissionsspektrums eignen sich folgende Einstellungen, die während der gesamten Messreihe unverändert bleiben müssen:
- Integrationszeit: 20 ms
- Scans zur Mittelwertbildung: 40
- Boxcar Breite: 2
Aktivieren sie die Lichtquelle
 und starten die Messung.
und starten die Messung.
Aufnahme von Absorptionsspektren

Um Absorptionsspektren aufzunehmen muss die Faser wie im Bild dargestellt mit dem Probenhalter verbunden werden und ein Neutraldichtefilter (ND-Filter), wie abgebildet, in den Strahlengang gesetzt werden. Verwenden Sie für die Messung eine 2-Wege-Küvette.
Zunächst wird wie bei der Aufnahme von Emissionsspektren beschrieben vorhandene Graphen-Tabs geschlossen und ein neuer geöffnet. Folgende Messparameter werden gewählt:
- Integrationszeit: 10 ms
- Scans zur Mittelwertbildung: 100
- Boxcar Breite: 2
Füllen Sie die Küvette mit dem im Versuch verwendeten Lösungsmittel jedoch ohne die zu messende Probe. Zur Aufnahme eines Dunkelspektrums wird ein einzelnes Spektrum der Probe bei ausgeschalteter Lichtquelle aufgenommen (PlayPause). über den Button
![]() wird dieses Spektrum als Dunkelspektrum festgesetzt. Anschließend wird ein Lampenspektrum bei eingeschalteter Lichtquelle
aufgenommen und durch Anklicken des Buttons
wird dieses Spektrum als Dunkelspektrum festgesetzt. Anschließend wird ein Lampenspektrum bei eingeschalteter Lichtquelle
aufgenommen und durch Anklicken des Buttons
![]() als Referenzspektrum (I0 -> siehe Vorlesungsskript Prof. Hartschuh) festgelegt, wobei zubeachten ist, dass die maximale Countzahl von 65000 nicht erreicht wird. Jetzt kann auf Absorptionsmessung umgeschaltet werden
als Referenzspektrum (I0 -> siehe Vorlesungsskript Prof. Hartschuh) festgelegt, wobei zubeachten ist, dass die maximale Countzahl von 65000 nicht erreicht wird. Jetzt kann auf Absorptionsmessung umgeschaltet werden
![]() und die Einheit der y-Achse wechselt automatisch auf „Absorption (OD)“.
und die Einheit der y-Achse wechselt automatisch auf „Absorption (OD)“.
Aufnahme von Transmissionsspektren
Zur Aufnahme von Transmissionsspektren werden die gleichen Einstellungen und Referenzspektren wie bei Absorptionsspektren verwendet. Lediglich der Messmodus wird auf Transmission umgeschaltet
![]()
Speichern von Spektren
Jede Gruppe erstellt sich zunächst einen Gruppenordner im Dokumentenordner des Computers. In diesem Ordner werden alle Spektren und Trenddiagramme dieser Gruppe gespeichert. Ist ein gewünschtes Spektrum festgehalten (Pause oder Play/Pause) kann es durch den Schalter
![]() oberhalb des Graphen gespeichert werden (nicht über das Datei-Menü!). Im sich öffnenden Dialog
oberhalb des Graphen gespeichert werden (nicht über das Datei-Menü!). Im sich öffnenden Dialog
wird der Speicherort und der Dateiname angegeben. Wichtig ist die Einstellung des Datei-Typs „Tab getrennt, ohne Kopfzeile“, sonst kann das Spektrum anschließend von keinem externen Programm geöffnet werden.
Trend-Diagramm für Kinetiken
Ist bereits eine Absorptions- oder Fluoreszenzmessung wie vorher beschrieben konfiguriert, lässt sich die Änderung
in den Spektren auch zeitlich verfolgen und kann durch den Schalter
![]() oberhalb des Spektrums gespeichert werden. Dies wird dazu verwendet um z.B. die Änderung der Optischen Dichte am Absorptionsmaximum im zeitlichen Verlauf einer Entfärbungsreaktion zu beobachten.
oberhalb des Spektrums gespeichert werden. Dies wird dazu verwendet um z.B. die Änderung der Optischen Dichte am Absorptionsmaximum im zeitlichen Verlauf einer Entfärbungsreaktion zu beobachten.
Im sich öffnenden Dialog
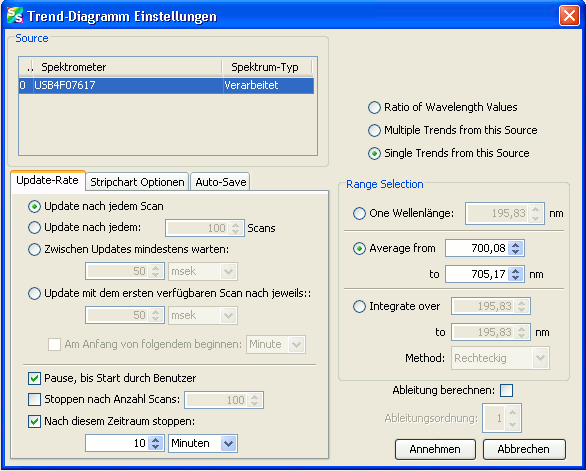
sind die Einstellungen wie abgebildet zu treffen. Lediglich die Angabe des Zeitraumes und der Bereichsauswahl sind abhängig vom jeweiligen Experiment. In der Bereichsauswahl wird der Bereich im Spektrum festgelegt, dessen zeitliche Änderung beobachtet werden soll (meist ist das der Bereich eines Absorptions- oder Emissionsmaximums). Die angegebenen Zahlen hier sind nur Beispiele!
Besonders wichtig ist auch noch die Eingabe der Auto-Save Optionen:
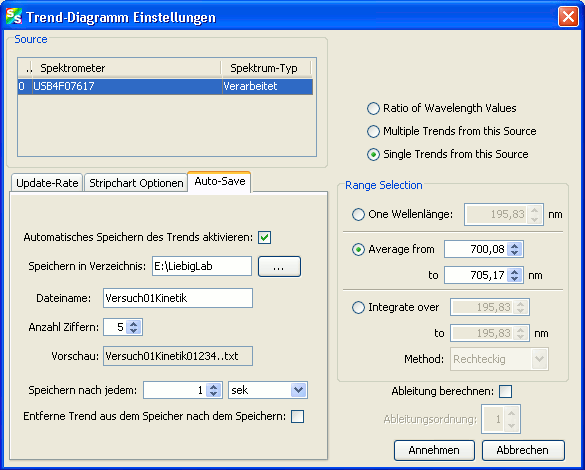
Aktivieren Sie das automatische Speichern, geben Sie den Speicherort und den Dateinamen an und geben Sie an, dass nach jeder Sekunde gespeichert werden soll. Nachdem alle Einstellungen getroffen wurden bestätigen Sie mit „Annehmen“. Darauf öffnet sich im Hintergrund ein weiterer Plot indem die Daten des Trend-Diagramms enthalten sind. Das im Vordergrund geöffnete Fenster kann geschlossen werden. Sollte eine Veränderung der Einstellungen nötig sein lässt sich diese Fenster jederzeit wieder über den Schalter „Konfigurieren“ aufrufen.
Sobald nun die Messung der Spektren im Hintergrund durch den Schalter
gestartet wurde lässt sich die Aufnahme des Trends durch den kleiner Play-Schalter starten. Die dargestellten Daten werden automatisch im Hintergrund in die von Ihnen vorher benannte Datei gespeichert. Die Aufnahme des Trends kann durch den Schalter
![]() gestoppt werden und durch den Schalter
gestoppt werden und durch den Schalter
![]() zurückgesetzt werden um einen weiteren Trend mit der fortlaufenden Nummerierung am Dateiende aufzunehmen.
zurückgesetzt werden um einen weiteren Trend mit der fortlaufenden Nummerierung am Dateiende aufzunehmen.
FAQ
Manchmal erkennt der Rechner das Spektrometer nicht. Schritte zur Lösung dieses Problems:
• Programm neustarten
• Alle Anschlüsse überprüfen, USB-Kabel an beiden Anschlüssen ein- und ausstecken
• Rechner neustarten
Es sind keine vernünftigen Spektren zu erkennen:
• OD-Filter bei Absorption eingebaut? bzw. bei Fluoreszens ausgebaut
• Lampe angeschaltet/angesteckt?
• Küvette steht nicht gerade im Küvettenhalter?
Absorptionsspektren zeigen keinen sinvolle Absorptionskurve mit einem eindeutigen Maximum:
• Konzentration der betrachteten Farbstofflösung ist zu hoch. (Absorptionsmaximum liegt über > OD 1,5)
Der Kalkkreislauf
Das Münchner Trinkwasser ist „hart“, es ist kalkreich. Der gelöste Kalk stammt aus den Kalkalpen (der größte Teil des Münchner Wassers kommt aus dem Mangfalltal), dabei kann Wasser unmöglich die bei der Analyse der Wasserhärte gefundenen Kalkmengen durch einfaches Auflösen aufgenommen haben. Wäre Kalk so gut wasserlöslich, wie es die recht hohe Menge in einem harten Trinkwasser, vor allem aber in manchen Mineralwässern, glauben macht, wäre die Lage von Muscheln mit ihrem äußeren Kalkskelett desolat. Hinter dem natürlichen Kreislauf des Kalks steckt ganz offensichtlich mehr, und zwar Säure-Base-Chemie.
Wissenswertes vorab
Calcit und Aragonit, die wichtigsten Calciumcarbonat-Modifikationen: Calciumcarbonat (Kalk) ist als Calcit und Aragonit eines der häufigsten Minerale der Erdkruste. Calcit kommt als Kalkstein, porös als Kreide, oder, nach Einwirkung von Druck, als Marmor vor. Im ebenfalls gebirgsbildenden Mineral Dolomit sind die Calcium-Ionen des Calcits zur Hälfte durch Magnesium-Ionen ersetzt. Im menschlichen Organismus kommt Calcit als Biomineral im Otoconium („Ohrsand“) im Gleichgewichtsorgan vor, die viel größeren Otolithe („Ohrsteine“) von Fischen bestehen aus Calcit und/oder Aragonit, während im Perlmutt der Muschelschale Calcit und Aragonit miteinander abwechseln. Die größte biogene Kalkmenge liegt in den Exoskeletten von Kalkalgen vor.
Kalk schmilzt beim Erhitzen nicht, er zersetzt sich: In der Technik findet Calciumcarbonat schon seit vorchristlicher Zeit (ca. 1500 v. Chr.) als Rohstoff für Mörtel Anwendung im Bauwesen. Grundlage dieser Verwendung ist das „Kalkbrennen“ und „Kalklöschen“, dem das „Abbinden“ und „Aushärten“ nach der Anwendung folgt. Calcit dient als „Marmor“ seit Jahrtausenden als Baustoff und Werkstoff für unzählige Kunstwerke. Bei der Erhaltung dieser Bau- und Kunstwerke ist die Reaktivität des Kalks gegenüber Säure das Hauptproblem: saurer Regen greift Marmor stark an und führt zu dessen „Verwitterung“.
Der größte Teil der Wasserhärte geht beim Erhitzen in Kalkabscheidungen über: Calciumcarbonat fällt beim Erhitzen von hartem Trinkwasser aus, wobei die lästigen Kalkablagerungen in Wasserkochern, Kaffeemaschinen, etc. entstehen („Carbonathärte“, „temporäre Härte“). Calcium- und Magnesiumsalze, die beim Erhitzen des Trinkwassers unverändert bleiben, ergänzen die temporäre um die „permanente Härte“. Die Summe aus temporärer und permanenter Härte ist die Gesamthärte des Wassers. Während die Wasserhärte auf der einen Seite für die Ernährung bedeutsam ist, führt sie auf der anderen zum Ausfallen von „Kalkseifen“ bei der Textilwäsche. Hier ist die Enthärtung des Wassers notwendig, die durch Ionenaustausch oder Komplexierung der Calcium- und Magnesium-Ionen gelingt.
Lernziele und einführende Literatur
Lernziele: Protolyse schwacher Säuren und Basen, Acidimetrie und Alkalimetrie, Kohlensäure, Löslichkeitsprodukt, Chelatkomplexe, Komplexometrie, Kalk und Kalkkreislauf, Wasserhärte, Gesamthärte und Carbonathärte.
Einführende Literatur: Mor9-486K28.9, Mor10-494K29.7, sowie Allgemeines zu Säuren und Basen im Mortimer.
Vorversuche
Salze entstehen bei der Reaktion von Säuren mit Basen in oft wässriger Lösung. Dabei kann die Säure und/oder die Base durch ihr Anhydrid ersetzt sein. Umgekehrt lassen sich Salze oft in Säure- und Baseanhydrid zerlegen. Die Vorversuche sind den Bezügen Säure-Säureanhydrid und Base-Baseanhydrid gewidmet.
Brennen und Löschen von Kalk
Salze werden thermisch zersetzt, wenn eine der entstehenden Komponenten bei der angewendeten Temperatur flüchtig ist. „Komponenten“ sind Base- und Säureanhydrid, falls ein Salz auf dem Papier aus diesen formuliert werden kann. Calciumcarbonat ist ein Beispiel: CaCO3 ⇌ CaO + CO2.
Etwas Calciumcarbonat wird auf einer Magnesiarinne mit dem Bunsenbrenner für einige Minuten auf Weißglut erhitzt. Nach Abkühlen wird der so erhaltene „Branntkalk“ (Calciumoxid) im nächsten Versuch verwendet.
Was erwarten Sie, wenn anstelle von CaCO3 das homologe CaSiO3 gebrannt wird?
Base- und Säureanhydride reagieren oft (Calciumoxid, Phosphorpentaoxid, Schwefeltrioxid), aber nicht immer (Magnesiumoxid, Siliciumdioxid) bereits unter Umgebungsbedingungen mit Wasser zum Hydroxid oder zur Säure. Bei Calciumoxid klappt es, der Vorgang heißt in der Technik „Kalklöschen“.
Der im vorherigen Versuch gebrannte Kalk wird in ein kleines Becherglas gegeben und vorsichtig mit Wasser versetzt. Dabei kommt es zu einer starken Wärmeentwicklung, die bei tropfenweiser Zugabe des Wasser so groß ist, dass ein Teil des Wassers sofort verdampft. Man erhält zunächst einen steifen Brei von Calciumhydroxid („Löschkalk“). Bei weiterer Zugabe von Wasser bildet sich eine milchige Flüssigkeit. Die so erhaltene „Kalkmilch“ wird filtriert und der pH-Wert des Filtrats mit einem Indikatorpapier getestet. Das Filtrat wird im nächsten Versuch weiter verwendet.
Formulieren Sie die Reaktionsgleichungen für das Kalklöschen.
Woran könnte es liegen, dass das homologe Magnesiumoxid, das als Magnesia zum Trockenhalten der Hände beim Geräteturnen und beim Felsklettern dient, keine alkalische Reaktion in Wasser hervorruft?
Reaktion von Calciumhydroxid mit Kohlendioxid
Bei dem Versuch finden nacheinander zwei wichtige Reaktionen statt: (1) Die erste Reaktion, die Entstehung von Kalk aus Löschkalk und Kohlendioxid, beschreibt das „Erhärten“ von Kalkmörtel. (2) Das anschließende Auflösen des entstandenen Calciumcarbonats in kohlendioxidhaltigem Wasser sowie die Umkehrung der Reaktion ist die Grundlage des Kalkkreislaufs der Natur.
Durch die im vorherigen Versuch hergestellte Lösung von Calciumhydroxid wird vorsichtig Atemluft durchgeblasen. Anschließend wird kurzzeitig erhitzt und das entweichende Kohlendioxid mit einem Tropfen Bariumhydroxid-Lösung, der an einem Glasstab über die Flüssigkeitsoberfläche gehalten wird, nachgewiesen.
Formulieren Sie die Reaktionsgleichungen für die einzelnen Schritte.
Das anfängliche Erhärten von Kalkmörtel wird gefördert, wenn in einem Neubau offene Holzfeuer unterhalten werden. Warum wirkt das besser als eine elektrische Beheizung?
Stalagmiten und Stalaktiten entstehen in einer Tropfsteinhöhle im Sinne des zweiten Versuchsteils. In einer Tropfsteinhöhle wird jedoch nichts erhitzt. Wieso kommt es trotzdem zur Reaktion?
Neutralisierende Wirkung von Leitungswasser
Der wichtigste mineralische Bestandteil von Leitungswasser ist Calciumhydrogencarbonat, das sowohl mit Säuren als auch mit Basen reagiert. Trinkwasser zeigt daher sowohl bei Säure- als auch bei Basezusatz ein neutralisierendes Verhalten.
Zwei von vier 200-mL-Bechergläsern werden zur Hälfte mit entmineralisiertem Wasser gefüllt, die beiden anderen mit Leitungswasser. Zu jeder Probe wird gleich viel Universalindikator-Lösung hinzugefügt. Nun gibt man zu je einer Probe des entmineralisierten Wassers und des Leitungswassers zwei Tropfen 1 m NaOH, zu den beiden anderen zwei Tropfen 1 m HCl. Notieren Sie die vom Indikator angezeigten pH-Werte.
Formulieren Sie die Gleichungen für die beiden Reaktionen, die den Säure- und den Baseverbrauch im Leitungswasser beschreiben.
Münchner Trinkwasser enthält im Mittel 5 mmol L-1 Hydrogencarbonat-Ionen. Rechnen Sie aus, wieviel Tropfen 1 m HCl nötig sind, um alles Hydrogencarbonat zu Kohlensäure zu protonieren (1 Tropfen = 0,05 mL).
Übungsanalysen
Bestimmung von Calcium
Die Bestimmung des Calciumgehalts erfolgt durch komplexometrische Titration mit Ethylendiamintetraacetat (edta). Die Methode kann nach Anpassung von pH-Bereich und Indikator zur quantitativen Bestimmung vieler Metall-Ionen verwendet werden.
Analyse 3: Calciumbestimmung
Zur Bestimmung des Calciumgehalts wird eine 50-mL-Probe mit 50 mL Wasser verdünnt. Anschließend wird eine Indikatorpuffertablette zugegeben. Nach deren Auflösen werden 2–3 mL 25 %ige Ammoniaklösung zugefügt und mit 0,1 m edta-Lösung von rot über grau nach grün titriert. Gehen sie zur Berechnung des Calciumgehalts davon aus, dass Calcium-Ionen und edta im gleichen Molverhältnis miteinander reagieren.
Formulieren Sie die Reaktionsgleichung für die Umsetzung von Calcium-Ionen mit edta-Lösung.
Die Titration wird in so stark alkalischer Lösung ausgeführt, dass Magnesium anstelle eines edta-Komplexes ein schwerlösliches Hydroxid bildet. Erwarten Sie, dass in Abwesenheit von Magnesium der Calciumgehalt auch in saurer Lösung bestimmt werden kann?
Vollanalyse: Wasser
Die „Härtebildner“ Magnesium und Calcium kommen als Hydrogencarbonat und Sulfat im Trinkwasser vor und bestimmen dessen Verwendbarkeit: so ist ein hoher Härtegrad für die Ernährung erwünscht, führt aber zu technischen Problemen durch die unerwünschte Bildung unlöslicher Calciumverbindungen (Kalk, „Kalkseife“, Calciumsalze von Säuren in Nahrungsmitteln, zum Beispiel bei Tee). Der Gehalt an Härtebildnern ist daher eine wichtige Kennzahl, die von Wasserwerken publiziert wird.
Bestimmung der Gesamthärte
Zur Bestimmung der Gesamthärte wird zunächst eine 0,01 m edta-Lösung hergestellt. Mit dieser wird anschließend die Probelösung titriert.
Zur Herstellung einer 0,01 m edta-Maßlösung wird eine bereitgestellte 0,1 m edta-Lösung (wässrige Lösung von Dinatrium-ethylendiammoniumtetraacetat, Na2H2edta) verdünnt. Mit Hilfe einer Vollpipette werden 25 mL der edta-Maßlösung in einen 250-mL-Messkolben gefüllt und dieser mit destilliertem Wasser bis zur Ringmarke aufgefüllt. Zur Bestimmung des Faktors werden ca. 100 mg Calciumcarbonat, welches zuvor bei 105 °C getrocknet wurde, eingewogen (genauen Wert notieren!). Das Calciumcarbonat wird in etwa 20 mL destilliertem Wasser aufgeschlämmt und mit 2 mL einer 2 m Salzsäure versetzt. Anschließend wird 5 Minuten zum Sieden erhitzt, die abgekühlte Lösung auf etwa 100 mL aufgefüllt und mit 1 m Natronlauge neutralisiert. Die Lösung wird dann in einen 250-mL-Messkolben überführt und dieser bis zur Ringmarke mit destilliertem Wasser aufgefüllt. Von dieser Lösung werden 25 mL in einen Erlenmeyerkolben pipettiert, mit destilliertem Wasser auf ca. 100 mL aufgefüllt und etwa 1 mL 6 m Natronlauge zugesetzt. Nach Zugabe von 5–7 Tropfen Calconcarbonsäure-Indikator wird mit der einzustellenden edta-Lösung von violett nach blau titriert. Der gesuchte Gehalt der edta-Lösung in mol L-1 ist dann gleich der Molzahl an Calcium in der abgemessenen Probe dividiert durch den Verbrauch an Maßlösung bei der Titration.
Die folgende Methode erfasst die Summe aus Calcium- und Magnesium-Ionen.
Analyse 4 (man.): Wasserhärte
Eine 25-mL-Probe des zu untersuchenden Trinkwassers wird auf 100 mL verdünnt und etwa 2 mL 25%iger Ammoniaklösung zugesetzt. Nach Zugabe einer Indikatorpuffertablette wird mit 0,01 m edta-Lösung bis zum Farbumschlag von rot über grau nach grün titriert.
Trotz europäischer Harmonisierungsbeschlüsse ist die Angabe der Wasserhärte in Grad Deutscher Härte (°dH) immer noch üblich. 1 °dH entspricht dabei der Summe der Magnesium- und Calcium-Ionen in 100 mL Wasser, ausgedrückt als mg Calciumoxid. In der Praxis: multiplizieren Sie die Millimolzahl Magnesium und Calcium in 100 mL Wasser mit der Molmasse von Calciumoxid (M = 56 g mol-1), um die Gesamthärte Ihrer Analysenlösung in °dH anzugeben.
Überprüfung der Gesamthärte
Um das oben erhaltene Ergebnis zu überprüfen, wird die Gesamthärte des zu untersuchende Trinkwasser noch einmal automatisch mit Hilfe des Titrierautomaten bestimmt. Die erhaltene Titrationskurve sollte folgendermaßen aussehen:
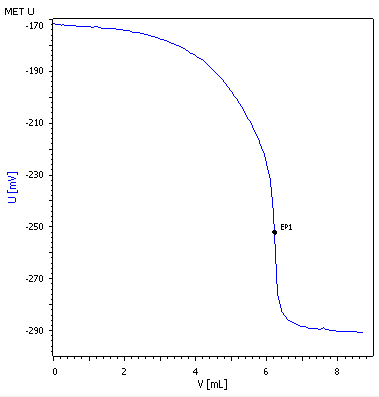
Analyse 4 (aut.): Wasserhärte (Kupferelektrode + Bezugselektrode)
Eine 25-mL-Probe des zu untersuchenden Trinkwassers wird in ein Becherglas gegeben und mit 20 mL Ammoniak/Ammoniumchlorid-Pufferlösung sowie 2 mL einer 0,05 m Kupfer(II)-edta-Lösung versetzt. Anschließend wird mit destilliertem Wasser auf 100 mL aufgefüllt. Die Methode „Titration mit 0,01 M EDTA-Lösung“ wird ausgewählt, die Kupfer- und die Bezugselektrode, sowie der Dosierer in die Probelösung getaucht (Rührfisch nicht vergessen!) und die Titration gestartet. Der Endpunkt sollte bei Spannungswerten ab ca. -220 mV erscheinen.
Die Methode ist für Wasserhärten über 10 °dH geeignet. Liegt der manuell ermittelte Härtegrad darunter, werden 50 mL des zu untersuchenden Wassers eingesetzt. Die Probe ist hierbei ebenso mit destilliertem Wasser auf 100 mL aufzufüllen.
Bestimmung der Gesamtalkalität
Die Gesamtalkalität erfasst alle Bestandteile eines Brauch- oder Trinkwassers, die oberhalb des Umschlagspunktes von Methylorange zugesetzte Säure binden. Die Gesamtalkalität wird daher auch als Methylorange-Alkalität bezeichnet.
Analyse 5 (man.): Gesamtalkalität
Für die Bestimmung der Gesamtalkalität werden 100 mL des zu untersuchenden Trinkwassers in einen Erlenmeyerkolben pipettiert. Statt Methylorange werden dem Wasser 4 Tropfen Methylrot und 15 Tropfen Bromkresolgrün zugesetzt, da der Umschlag dieses Mischindikators leichter erkennbar ist, als der des Methylorange-Indikators. Titrieren sie nun mit einer bereitgestellten 0,1 m HCl-Maßlösung von türkis bis zum Farbumschlag nach rot (mit leichtem Graustich). Kurz bevor der Indikator nach rot/grau umschlägt, erhält man als Zwischenstufe eine graue Lösung. Geben Sie den Säureverbrauch in mmol L-1 an.
Überprüfung der Gesamtalkalität
Um das oben erhaltene Ergebnis zu überprüfen, wird die Gesamtalkalität des zu untersuchenden Trinkwassers noch einmal automatisch mit Hilfe des Titrierautomaten bestimmt. Lesen Sie die verbrauchte Säuremenge auf dem Plateau nach dem Steilanstieg ab (die Titration stoppt nach Erreichen des auf 4,3 eingestellten pH-Wertes). Geben Sie den Säureverbrauch in mmol L-1 an. Die erhaltene Titrationskurve sollte folgendermaßen aussehen:
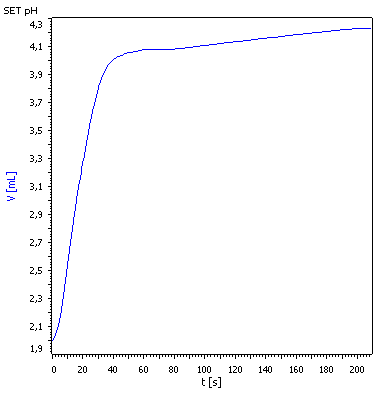
Analyse 5 (aut.): Gesamtalkalität (pH-Glaselektrode)
Eine 100-mL-Probe des zu untersuchenden Trinkwassers wird in ein Becherglas gegeben. Die Methode „Gesamtalkalität mit 0,1 m HCl“ wird ausgewählt, die pH-Elektrode und der Dosierschlauch in die Probelösung getaucht (Rührfisch nicht vergessen!) und die Titration gestartet. Berechnen Sie den Säureverbrauch in mmol L-1.
Welche Trinkwasserbestandteile führen zu einem Säureverbrauch (Reaktionsgleichungen)?
Diese allgemeinen Konzepte sollten Sie hinter den Versuchen erkennen …
Die Salzbildung aus Metallhydroxid und Säure, Metallhydroxid und Säureanhydrid, Metalloxid und Säure, oder aus Metalloxid und Säureanhydrid, ist eine ebenso grundlegende Gruppe von Reaktionen wie die Umsetzungen von Salzen mit einer Komponente aus demselben Katalog (Säure, Säureanhydrid, Base, Baseanhydrid) unter Verdrängung eines schwächeren oder entfernbaren Bindungspartners. Wird CaCO3 („CaO·CO2“) bei ausreichend hohen Temperaturen mit SiO2 erhitzt, so wird das leichtflüchtige Säureanhydrid CO2 vom nicht flüchtigen Säureanhydrid SiO2 aus dessen Salz verdrängt (formulieren Sie die Gleichung). Dieses Prinzip wird bei der industriellen Synthese von Zement angewandt.
Ein Säureanhydrid wie SO2 wird durch Ca(OH)2 (und Sauerstoff) gebunden. Dies wird zur Abtrennung des bei der Verbrennung schwefelhaltiger fossiler Brennstoffe entstehenden umweltschädlichen Schwefeldioxids aus Kraftwerkabgasen genutzt (Rauchgasentschwefelung) – es wird „Chemiegips“, CaSO4, erhalten.
(1) Skizzieren Sie den natürlichen Kalkkreislauf. (2) Beschreiben Sie, wie es zur Bildung von Tropfsteinhöhlen kommt. (3) Beschreiben Sie, wie die Carbonathärte in unser Trinkwasser kommt. (4) Beschreiben Sie, wo geographisch gesehen hartes, wo weiches Wasser vorkommt.
… noch ein paar Tipps, wie Sie Fehler vermeiden können:
• Lesen Sie die Versuchsvorschriften aufmerksam und vollständig durch, alle nötigen Informationen sind darin enthalten.
• Überprüfen Sie zu Beginn des Praktikums Ihre Glasgeräte auf ihre Sauberkeit. Sind Ihre Messgefäße staub- und fettfrei?
• Machen Sie sich noch einmal mit dem Inhalt des Kapitels 4 (Maßanalyse) aus dem Vorkurs vertraut.
• Verwenden Sie zum Verdünnen Ihrer Lösung ausschließlich vollentsalztes („destilliertes“) Wasser.
• Stellen Sie sich gegebenenfalls Vergleichslösungen her, um den Umschlagspunkt bei den Titrationen besser erkennen zu können.
Säure und Base und Chelatligand: Aminosäuren als polyfunktionelle Moleküle
Aminosäuren sind nicht nur die Bausteine der Peptide und Proteine, Aminosäuren selbst sind auch ohne die Einbindung in ein Protein ungewöhnliche Moleküle – sie sind „polyfunktionell“. Während alle Aminosäuren eine Carboxylfunktion und eine Aminofunktion tragen, weisen einige der 20 proteinogenen Aminosäuren weitere funktionelle Gruppen in ihrer Seitenkette auf. So treten dort weitere Aminogruppen und Carboxylgruppen auf, aber auch Hydroxyl- und Thiolgruppen, sowie komplexere Strukturelemente wie Imidazol-, Phenol- oder Indolringe. In diesem Projekt lernen Sie, die Aminosäuren zu unterscheiden und ein Aminosäuregemisch aufzutrennen. Ein besonderer Schwerpunkt: Aminosäuren als polyfunktionelle Moleküle, deren basische Funktionen nicht nur mit der Lewissäure H+, sondern auch mit Lewis-sauren Metallionen reagieren können.
Wissenswertes vorab
Ampholyte
Aminosäuren sind Ampholyte. Deren wesentliche Kennzahlen können einer Titrationskurve entnommen werden. Eine Möglichkeit, eine alle Protolyseschritte umfassende Kurve zu erhalten, ist in der folgenden Abbildung genutzt worden. Diese zeigt die Titrationskurve der einfachsten Aminosäure, Glycin (Aminoessigsäure, H2NCH2COOH). Als Ampholyt liegt Glycin nach dem Auflösen in Wasser so vor, dass die stärkste saure Funktion (die COOH-Gruppe) deprotoniert und die stärkste basische Funktion (die NH2-Funktion) protoniert ist. Es liegt also das Zwitterion H3N+CH2COO− vor, das durch die Acidität der Ammoniumfunktion (pKS2) und die Basizität der Carboxylat-Funktion (pKB = 14 − pKS1 mit pKS1 als der Säurekonstante der zu Carboxylat konjugierten Säure, der COOH-Funktion) gekennzeichnet ist.
Die Probe wurde nun vorbereitet, indem bis zu einem recht hohen pH-Wert von ca. 12,5 Natronlauge zugefügt wurde. Bei diesem pH-Wert sind alle sauren Funktionen einer Aminosäure deprotoniert. Glycin liegt unter diesen Bedingungen als Glycinat-Monoanion (Aminoacetat, H2NCH2COO−) vor. Bei der anschließenden Titration mit 1 m Salzsäure wird bis zum Punkt EP1 nur die überschüssige Natronlauge verbraucht. EP1 markiert daher den Startpunkt der eigentlichen Glycinat-Titration, den Titrationsgrad 0 (τ = 0).
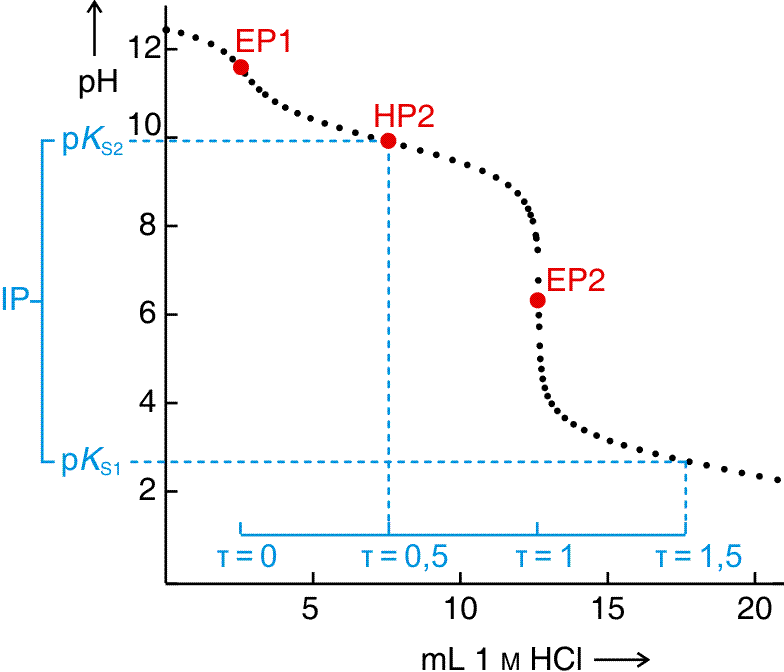
Titration von 755 mg Glycin (nach Zugabe von NaOH bis zum Erreichen von pH 12,5) mit 1 m HCl. Rot: Markierungen im Messprotokoll, blau: siehe Text.
Bei τ = 0,5 ist der erste Pufferpunkt erreicht (im Messprotokoll HP2), bei dem gleiche Anteile Glycinat und Glycin vorliegen. Dabei ist die Hälfte des Glycinats an seiner basischsten Position, der Aminogruppe, protoniert worden – aus der Hälfte des Glycinats ist das Zwitterion H3N+CH2COO− entstanden. Bei HP2 wird auf der Ordinate der pKS2-Wert abgelesen, der Säurekonstante der Ammoniumfunktion (9,89; vgl. 9,78 in Voet & Voet, Biochemistry).
Die weitere Titration führt zu EP2. Hier ist alles Glycinat protoniert, so dass nur noch das Zwitterion vorliegt – der isoelektrische Punkt ist erreicht. Bis hierher wurden 1 mol Protonen zu 1 mol Glycinat zugefügt, der Titrationsgrad ist 1 (τ = 1). Wurde die Aminosäure eingewogen, so ergibt der Quotient aus der Einwaage in mg und der zugegebenen Menge Protonen in mmol die Molekülmasse der Aminosäure. Da bei Glycin an diesem Äquivalenzpunkt das Zwitterion vorliegt, liegt hier auch der isoelektrische Punkt (IP) des Glycins, auf dessen Bestimmung weiter unter eingegangen wird.
Bei der weiteren Titration wird ein zweiter Pufferpunkt und ein zweiter Äquivalenzpunkt erwartet. Im Vergleich mit einem verwandten Ampholyten wie Ammoniumacetat sind die meisten Aminosäuren aber so sauer, dass die Carboxylfunktion bei den üblichen Konzentrationen durch die zugesetzte Salzsäure nicht mehr protoniert wird und bei τ = 2 kein Wendepunkt mehr zu erkennen ist. Aus diesem Grund ist auch die Bestimmung von pKS1, der Säurekonstanten der Carboxylfunktion, beim zweiten Pufferpunkt (τ = 1,5) mit einer höheren Unsicherheit behaftet als die Bestimmung von pKS2. Im Beispiel ergibt sich pKS1, der pH-Wert bei τ = 1,5, zu 2,66 (vgl. 2,35 in Voet & Voet, Biochemistry).
Der isoelektrische Punkt ergibt sich für Glycin nach der Bestimmung von pKS1 und pKS2 als Mittelwert der beiden Säurekonstanten zu 6,3. (Setzen Sie in der Praxis IP und EP2 nicht gleich, da EP2 in diesem steilsten Kurvenabschnitt einen besonders großen Fehler des Ordinatenwerts aufweist! Bestimmen Sie also den IP nur als Mittelwert der Säurekonstanten.)
Warum ist der pKS1-Wert des Glycins so deutlich kleiner als der von Essigsäure mit einem pKS-Wert von 4,75?
Der isoelektrische Punkt (IP)
Definitionen: Wie schon zuvor sei pKS1 der pKS-Wert der Carboxylfunktion, pKS2 der pKS-Wert der zur Aminofunktion konjugierten Säure; pKS3 sei nun der pKS-Wert der zweiten Säurefunktion im Fall einer sauren Seitenkette oder der pKS-Wert der konjugierten Säure im Fall einer basischen Seitenkette.
Der IP einer Aminosäure ergibt sich als der pH-Wert, bei dem die Aminosäure als Zwitterion vorliegt. Dieser pH-Wert wird dadurch bestimmt, welche funktionelle Gruppe des Ampholyten gegenüber Wasser als Säure und welche als Base wirkt.
Enthält die Aminosäure keine protolysierende Seitenkette, so liegt der Ampholyt hauptsächlich mit deprotonierter Carboxylfunktion und protonierter Aminofunktion vor (der Fall entspricht also dem Glycin-Beispiel). Die Säurewirkung gegenüber Wasser beruht auf der protonierten Aminofunktion (pKS2), die Basewirkung auf der Carboxylatfunktion (pKS1), der IP ergibt sich als
pH = ½ (pKS1 + pKS2).
Besitzt die Aminosäure eine saure Seitenkette, wird zuerst von dem üblichen Zwitterion ausgegangen. Dessen Basizität gegenüber Wasser beruht auf der Carboxylatfunktion (pKS1), seine Acidität aber beruht auf der Seitenkette (pKS3). Der IP ist nun
pH = ½ (pKS1 + pKS3).
Läge der Fall vor, dass pKS1 größer als pKS3 ist, würde sich der Ausgangspunkt der Ableitung ändern, nicht aber das Ergebnis.
Auch wenn die Aminosäure eine basische Seitenkette besitzt, wird zuerst von dem üblichen Zwitterion ausgegangen. Dessen Acidität gegenüber Wasser beruht auf der protonierten Aminofunktion (pKS2), seine Basizität beruht nun aber nicht auf der Carboxylatfunktion, sondern auf der basischen Seitenkette (pKS3). Der IP ist nun
pH = ½ (pKS2 + pKS3).
Ist die Basizität der Seitenkette größer als die der Aminofunktion, so ändert sich auch hier nur der Ausgangspunkt der Ableitung, aber nicht das Ergebnis.
Beispiel 1: Phenylalanin. pKS1 = 2,20, pKS2 = 9,31. Der IP ist:
pH = ½ (pKS1 + pKS2) = ½ (2,20 + 9,31) = 5,76
Beispiel 2: Glutaminsäure mit einer sauren Carboxylseitenkette. pKS1 = 2,10, pKS2 = 9,47, pKS3 = 4,07. Der IP errechnet sich zu:
pH = ½ (pKS1 + pKS3) = ½ (2,10 + 4,07) = 3,09
Beispiel 3: Histidin mit einer basischen Imidazolseitenkette. pKS1 = 1,80, pKS2 = 9,33, pKS3 = 6,04. Der IP errechnet sich zu:
pH = ½ (pKS2 + pKS3) = ½ (6,04 + 9,33) = 7,69
Lernziele und einführende Literatur
Lernziele: (Dünnschichtchromatographie), Aminosäuren als Ampholyte, isoelektronischer Punkt, Aminosäuren als Chelatliganden.
Einführende Literatur: Mor-605K33.6, Mor-618K33.10, Mor-302K18.4
Vorversuche
Die Chromatographie ist ein Verfahren zum Auftrennen eines Stoffgemisches in seine Einzelbestandteile mit Hilfe einer stationären und einer mobilen Phase. Dieses qualitative Verfahren ermöglicht es auch, ein Gemisch aus Aminosäuren aufzutrennen. Dabei werden die unterschiedlichen Wanderungsgeschwindigkeiten der einzelnen Aminosäuren im Fließmittel ausgenutzt, um beispielsweise die Zusammensetzung eines Proteins zu analysieren.
Dünnschichtchromatographie von Aminosäuren
Bei der Chromatograpie wird die unterschiedliche Wanderungsgeschwindigkeit verschiedener Moleküle und damit das Verteilungsgleichgewicht eines Stoffes zwischen zwei Phasen – einer stationären und einer mobilen Phase – zur Stofftrennung genutzt. Bei der Dünnschichtchromatographie ist die stationäre Phase eine wenige Zehntel Millimeter dünne Schicht aus feinkörnigem Aluminiumoxid oder Siliciumdioxid. Entscheidend für die Identifizierung der einzelnen Aminosäuren ist ihr Retentionsfaktor (Rf-Wert) unter den jeweiligen Versuchsbedingungen. Der Rf-Wert ist der Quotient aus der Laufstrecke der Substanz und der Laufstrecke der Fließmittelfront vom Startpunkt aus; er ist für ein vorgegebenes chromatographisches System für eine Verbindung charakteristisch. Verschiedene Aminosäuren unterscheiden sich in ihrem Retentionsfaktor, wodurch die Analyse eines Aminosäuregemisches möglich ist.
Lösen Sie zur Herstellung einer Ninhydrin-Lösung 1,5 g Ninhydrin in einem Gemisch aus 5 mL konzentrierter Essigsäure und 500 mL 95%-igem Ethanol. Eine Lösung pro Praktikumssaal ist ausreichend!
Geben Sie in eine Chromatographiekammer ein Gemisch aus n-Butan-1-ol, Eisessig und Wasser im Volumenverhältnis 4:1:1, so dass die Kammer ca. 1 cm hoch gefüllt ist, stellen Sie ein passendes Stück Filterpapier hinzu und verschließen Sie die Kammer, um den Luftraum mit dem Lösungsmittelgemisch zu sättigen.
Ziehen Sie vorsichtig mit einem weichen Bleistift auf der DC-Platte 1 cm von der kürzeren Kante entfernt einen zur Kante parallelen Strich als Startlinie und unterteilen Sie den Strich in 1-cm-Abschnitte. Auf die Startlinie werden die Lösungen der reinen Aminosäuren (Arginin, Alanin, Histidin, Lysin, Methionin, Phenylalanin, Prolin, Valin) und das zu bestimmende gelöste Gemisch mit 1 cm Abstand untereinander und auch zum Rand mit einer feinen Kapillare aufgetragen. Tauchen Sie hierzu eine saubere Kapillare in die entsprechende Lösung, worauf durch Kapillarkräfte eine ausreichende Menge der Lösung in der Kapillare aufsteigt. Die Spitze der Kapillare wird nun ruhig und senkrecht auf die vorgezeichnete Stelle aufgesetzt. Man lässt einen kleinen Teil der Lösung ausfließen. Der gebildete Fleck wird kurz getrocknet. Dabei wird darauf geachtet, dass der Durchmesser des Flecks nicht größer als 2–3 mm wird.
Stellen Sie nun die DC-Platte in die Chromatographiekammer (die Sie wegen der Laufmittelsättigung nicht unnötig offen stehen lassen!) und nehmen Sie sie erst dann heraus, wenn sich die Fließmittelfront ca. 2 cm vom oberen Rand entfernt befindet. Markieren Sie diesen Rand der Fließmittelfront und lassen die Platte trocknen. Im Anschluss besprühen Sie die Platten vorsichtig mit einer Ninhydrin-Lösung und lassen erneut trocknen. Messen Sie nun den Abstand von der Mitte eines jeden Farbflecks zur Startlinie und berechnen Sie den Rf-Wert, indem Sie den Abstand durch den Abstand von der Startlinie bis zur Fließmittelfront teilen.
Welche Aminosäuren lagen in Ihrem Gemisch vor?
Protolysegleichgewichte bei Aminosäuren
Die Titration der Aminosäure-Zwitterionen zeigt deren Ampholytverhalten. Neben den Protolysekonstanten der einzelnen funktionellen Gruppen ist der isoelektrische Punkt eine typische Kenngröße, bei dem ein nach außen elektroneutrales Zwitterion vorliegt.
Die pKS-Werte in der folgenden Tabelle sind so definiert wie im Abschnitt „Wissenswertes“ beschrieben. R ist die Seitenkette an Cα.
| R | Mr | pKS1 | pKS2 | pKS3 | IP | ||
|---|---|---|---|---|---|---|---|
| Alanin | Ala | CH3 | 89,093 | 2,35 | 9,87 | – | 6,11 |
| Asparagin-Monohydrat | Asn | CH2CONH2 | 150,134 | 2,14 | 8,72 | – | 5,43 |
| Asparagins. | Asp | CH2COOH | 133,103 | 1,99 | 9,90 | 3,90 | 2,95 |
| Cystein | Cys | CH2SH | 121,159 | 1,92 | 10,70 | 8,37 | 5,15 |
| Glutamins. | Glu | CH2CH2COOH | 147,129 | 2,10 | 9,47 | 4,07 | 3,09 |
| Histidin | His | CH2C3H3N2 | 155,155 | 1,80 | 9,33 | 6,04 | 7,69 |
| Threonin | Thr | CH(CH3)OH | 119,119 | 2,09 | 9,10 | – | 5,60 |
(pH-Glaselektrode)
Ca. 1,3 g der Probe werden auf der Analysenwaage genau eingewogen, in 100 mL Wasser gelöst, mit 6 m Natronlauge auf einen pH-Wert von ca. 12,5 eingestellt (pH-Meter) und anschließend mit 1 m Salzsäure mittels Unitrode am Titrationsautomaten titriert.
Bestimmen Sie (1) den pKS2-Wert der Aminosäure aus Ihrer Titrationskurve und vergleichen Sie mit den tabellierten Werten, und (2) die Molmasse ihrer Probe aus der Einwaage und dem Verbrauch des am Besten sichtbaren Protonierungsschritts. Um welche Aminosäure handelt es sich?
Wie groß ist die prozentuale Abweichung der von Ihnen bestimmten Molmasse und dem Tabellenwert?
Bestimmen Sie aus Ihrer Titrationskurve näherungsweise den pKS1-Wert, indem Sie in sinnvoller Weise den Verbrauch des am Besten sichtbaren Protonierungsschritts zur Extrapolation ins Saure verwenden (siehe Muster).
Glycinato-Kupfer(II)-Komplexe: Titration von Glycin in Anwesenheit von Kupfernitrat
Die Anwesenheit von Metallkationen, die von Aminosäuren als Komplexliganden gebunden werden können, beeinflusst die Protolysegleichgewichte, da um die Basen (Amin, Carboxylat, deprotonierte Seitenkette) nun ein Wettbewerb zwischen dem Proton und dem Metallkation entsteht. Im ersten Versuchsteil titrieren Sie daher die reine Aminosäure in Abwesenheit von Metallkationen, wohingegen Sie im zweiten Versuchsteil als Metallkation Kupfer(II) vorliegen haben. Führen Sie den Versuch in Dreiergruppen aus.
(pH-Glaselektrode)
Titration von Glycin:
Glycin (2 mmol) wird in 99,0 mL 0,1 m KNO3-Lösung und 1,00 mL 1 m Salpetersäure gelöst. Stellen Sie den pH-Wert mit Hilfe von HCl auf 2,5 ein (pH-Meter) und titrieren Sie diese Lösung mit 0,5 m NaOH. Nehmen Sie eine Titrationskurve auf.
(pH-Glaselektrode)
Titration von Glycin in Anwesenheit von Kupfer(II):
Glycin (2 mmol) und Kupfer(II)-nitrat (1 mmol) werden in 99,0 mL 0,1 m KNO3-Lösung und 1,00 mL 1 m Salpetersäure gelöst. Stellen Sie den pH-Wert mit Hilfe von HCl auf 2,5 (pH-Meter) und titrieren Sie diese Lösung mit 0,5 m NaOH. Nehmen Sie eine Titrationskurve auf.
Interpretieren Sie die unterschiedlichen Kurvenverläufe. Dazu sollten Sie die beiden Titrationskurven in einem Diagramm darstellen. Gehen Sie in der Datenbank mit der Maus in das Fester mit der Titrationskurve und wählen Sie nach einem Rechtsklick den Punkt „Messpunktliste“ aus. Hier markieren Sie alle Werte und kopieren sie (zum Beispiel mit Strg+C). Fügen Sie die Werte beider Titrationskurven in ein Tabellenkalkulationsprogramm Ihrer Wahl ein und stellen Sie die beiden Kurven zusammen in einem Graphen dar. Welche Zusammensetzung ergibt sich für den Komplex, der aus Glycin und Kupfer-Ionen gebildet wird?
Wie könnte der Aufbau des aus der Titrationskurve abgeleiteten Komplexes aussehen?
Übungsanalyse
Titration einer Ammoniumacetat-Lösung
Im folgenden Versuch wird mit Ammoniumacetat experimentiert. Lernziel ist, die Parallelen zwischen diesem typische Ampholyt und einer Aminosäure zu erkennen. Die erhaltene Titrationskurve sollte folgendermaßen aussehen:
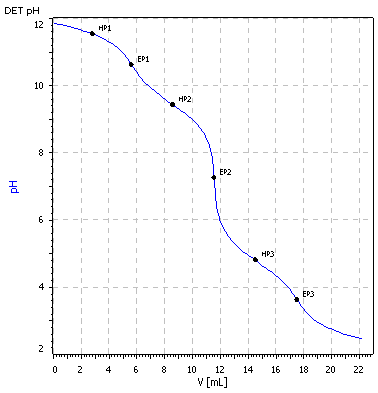
@ Analyse 6: Ammoniumacetat-Bestimmung (pH-Glaselektrode)
Eine 50 mL-Probe einer Ammoniumacetatlösung wird auf ca. 100 mL verdünnt, mit 1 m Natronlauge auf einen pH-Wert von ca. 11,5 eingestellt (pH-Meter) und sofort anschließend am Titrationsautomaten mit 0,1 m Salzsäure titriert.
Warum soll die Lösung nach der Laugezugabe nicht herumstehen? Woran erkennen Sie eine hierauf beruhende Störung?
Errechnen Sie den Ammoniumacetatgehalt der Analyselösung aus dem Verbrauch zwischen dem zweiten und dritten Äquivalenzpunkt.
Drucken Sie ihr Titrationsdiagramm aus und erklären Sie alle Wendepunkte. Welcher dieser Punkte wird erreicht, wenn Ammoniumacetat in reinem Wasser gelöst wird (keine Laugezugabe). Wie errechnet sich der pH-Wert an diesem Punkt?
Welche Parallelen zwischen der bereits durchgeführten Titration einer Aminosäure und der Titration von Ammoniumacetat sehen Sie?
Vergleichen Sie die von Ihnen ermittelten Werte der Aminosäure-Titration für pKS1 und pKS2 mit den Werten von Glycin (2,35; 9,78), der einfachsten Aminosäure, und den eben bestimmten pKS-Werten von Ammoniumacetat. Erklären Sie die Unterschiede.
Vollanalysen
Komplexometrische Bestimmung von Eisen mit edta
Ethylendiamintetraessigsäure ist eine synthetische Aminosäure mit besonders vielen funktionellen Gruppen, der Sie seltener in der Biochemie als in der analytischen Chemie begegnen. Das vierfach negagtiv geladene edta-Ion (Ethylendiamintetraacetat, Titriplex III), welches Sie bereits im Projekt „Kalkkreislauf“ kennengelernt haben, bildet mit Metallen Chelatkomplexe. Hier lernen Sie nun die Konkurrenz von Metall-Ionen und Protonen um die funktionellen Gruppen dieser polyfunktionellen Aminosäure kennen.
Analyse 7 (man.): Titrimetrische Eisen-Bestimmung
25 mL der Analysenlösung werden in einem Becherglas auf 100 mL verdünnt. Es werden 3–5 Tropfen Indikatorlösung (Sulfosalicylsäure, 5 g in 95 mL dest. H2O) zugegeben, worauf sich die Lösung rot-violett verfärbt, und mit 0,1 m edta bis zur ursprünglichen Gelbfärbung der Lösung (Vergleich: ausgegebene Lösung) titriert. Ein zu früher Endpunkt kann im Falle, dass zu wenig Indikator zugegeben wurde, vorgetäuscht werden. Es ist daher zweckmäßig, am Ende der Titration noch einige Tropfen Indikator zuzusetzen. Bei erneuter Rotfärbung wird weitertitriert.
Gravimetrische Metallbestimmung
Die Präzision moderner Analyseverfahren wird oft überschätzt. Moderne instrumentelle Methoden sind oft wesentlich schneller als alte Verfahren, aber keineswegs präziser. Ein Musterbeispiel ist die Gravimetrie, die sich im Hinblick auf die erreichbare Genauigkeit mit jeder anderen Methode messen kann. Der hohen erreichbaren Präzision steht eine hohe Anfälligkeit für experimentelle Fehler und ein hoher Zeitaufwand gegenüber. Man erreicht also nur dann beste Ergebnisse, wenn man alles richtig macht – also die optimale Methode, um präzises Arbeiten im Labor zu lernen.
Die Chemie ist übersichtlich: Zur Bestimmung eines Eisengehaltes wird zum Beispiel aus einer Eisen(III)-Salzlösung das schwerlösliche Hydroxid gefällt und durch eine kontrollierte Behandlung in die stabile Wägeform, Hämatit (α-Fe2O3), überführt. In einer ähnlichen Verfahrensweise kann auch Aluminium als α-Al2O3 bestimmt werden, alternativ als Oxinat. Hierzu werden Al3+-Ionen in essigsaurer, acetatgepufferter Lösung mit 8-Hydroxychinolin (Oxin, abgekürzt Hquin) versetzt. Dabei bildet sich ein schwerlöslicher Niederschlag des inneren Komplexsalzes Aluminiumoxinat, der nach dem Trocknen direkt zur Auswaage gebracht wird.
Analyse 8 (man.): Gravimetrische Aluminium-Bestimmung (als Aluminiumoxinat)
Bereiten Sie vor der Fällung als Waschflüssigkeit: 1 mL 2 m Essigsäure und 0,5 ml 2 m Natronlauge mit Wasser auf 100 mL verdünnen.
25 mL der Probelösung werden mit 25 mL Wasser in ein Becherglas gebracht und mit 2 m NaOH tropfenweise bis zur ersten Trübung versetzt (pH = 4–9). Danach werden 5 mL einer 2 m HCl zur Probe gegeben und mit Wasser auf 140 mL verdünnt. Nun lässt man aus einer Bürette unter Rühren langsam 40 mL einer 0,1 m Oxinacetatlösung einfließen. Die klare Lösung wird auf 75–85 °C erwärmt, und anschließend werden innerhalb von 10–15 Minuten langsam 15 mL 2 m Ammoniak als Pufferkomponente eingetropft. Beim Auftreten der ersten Trübung wird die Zugabe der Pufferlösung für zwei Minuten – jedoch unter weiterem Rühren – unterbrochen und der pH-Wert überprüft (Sollwert 4,2–4,8). Danach wird der Rest der Pufferlösung zugetropft. Der pH-Wert wird dabei stets überprüft, und bei Bedarf wird durch weitere Zugabe von Ammoniak auf den angestrebten Wert korrigiert. Die Lösung wird schließlich noch für 30 Minuten bei 60–70 °C gerührt. Nach Absetzen des grüngelben Feststoffes wird die Lösung mit dem Niederschlag heiß über einen G4-Glasfiltertiegel dekantierend filtriert. Zum Aufbringen der Niederschlagsreste wird das gelbe und wieder erwärmte Filtrat verwendet. Der Niederschlag wird mit 25 mL heißer (80–90 °C) Waschflüssigkeit und schließlich mit 5 mL heißem Wasser (durch Auftropfen mit einer Pipette!) gewaschen. Der Niederschlag wird zur Massekonstanz (mindestens zwei Stunden) bei 130 °C im Trockenschrank getrocknet. Nach Abkühlen des Tiegels im Exsikkator auf Raumtemperatur erfolgt die Auswaage.
Geben Sie die Formel des gebildeten Komplexsalzes an.
Skizzieren Sie die Lewisformel von H2edta2−, dem Anion des „EDTA-Dinatriumsalzes“, des Reagenzes zur Herstellung der Titrierflüssigkeit. Falls Ihr Reagenz als „Ethylendiamintetraessigsäure“ vorlag, skizzieren Sie deren Formel.
Stellen Sie die Gleichung für die Reaktion zwischen der Titrierflüssigkeit Na2H2edta und Fe3+ auf. Berechnen Sie die Menge an Fe3+ im 250-mL-Messkolben in mg.
Skizzieren Sie die räumliche Struktur eines Metall-edta-Komplexes.
Die komplexometrische Bestimmung von Mg2+ und Ca2+ mit edta erfolgt bei einem pH-Wert von 10, Calcium kann neben Magnesium bei einem pH-Wert von 12 bestimmt werden, da Magnesium bei diesem pH-Wert als Hydroxid vorliegt. Die quantitative Analyse sowohl von Magnesium als auch von Calcium erfolgt demgemäß im Alkalischen. Warum kann Fe3+ im Sauren bestimmt werden?
Diese allgemeinen Konzepte sollten Sie hinter den Versuchen erkennen …
Aminosäuren sind polyfunktionell. Es gibt kaum eine Stoffklasse, bei der relativ kleine Moleküle so unterschiedliche funktionelle Gruppen tragen. Das wesentliche Lernziel bei diesem Projekt ist es, die Protolysechemie der Aminosäuren als Folge des Zusammenspiels der unterschiedlichen funktionellen Gruppen zu verstehen. Vor allem die Frage nach dem isoelektrischen Punkt erfordert eine nähere Beschäftigung mit den Protolysekonstanten der verschiedenen Aminosäuren.
Erklären Sie den zwitterionischen Charakter von Aminosäuren.
„Aminosäurelösungen wirken als Puffer.“ – liest man schon mal so. Prüfen Sie diese Aussage anhand Ihrer Titrationskurven.
… noch ein paar Tipps, wie Sie Fehler vermeiden können:
zur Dünnschichtchromatograpie: Tüpfeln Sie möglichst wenig Substanz auf die DC-Platte.
zur Gravimetrie: Da Beschriftungen auf dem Tiegel nicht halten, sollte ein Ofenplan aufgestellt werden.
Bleiche, Desinfektion, oxidativer Stress: starke Oxidationsmittel
Unser Leben in einer Sauerstoffatmosphäre hat ihren Preis: wir sind reaktiven Sauerstoffspezies (ROS) ausgesetzt, die bei Stoffwechselvorgängen entstehen können. Zu ihnen gehören sowohl freie Radikale (das Hyperoxid-Radikal-Anion, O2•−, das Hydroxyl-Radikal, HO•, und das Hydroperoxid-Radikal, HOO•), als auch nichtradikalische reaktive Stoffe wie Wasserstoffperoxid, H2O2, Ozon, O3, oder Singulett-Sauerstoff. Aufgrund ihrer Schädlichkeit für den Organismus werden Sie durch Enzyme entgiftet, die gegen diesen „oxidativen Stress“ gerichtet sind – ein Stoff wie H2O2 wird aber auch gezielt durch Organismen synthetisiert und eingesetzt. Ein Beispiel ist die H2O2-Produktion durch holzabbauende Pilze. In gleicher Weise stellen diese Sauerstoffspezies aufgrund ihrer Reaktivität auch wichtige Reaktionspartner in der technischen und synthetischen Chemie dar. Ein wichtiger Zweig der technischen Chemie befasst sich mit der Produktion von Bleich- und Waschmitteln, in denen vor allem Peroxide und Chlor-freisetzende Verbindungen Verwendung finden.
Die O-O-Gruppierung („Peroxo“-Gruppe) kommt in vielen starken Oxidationsmitteln vor. Ein Beispiel für ein geläufiges starkes Oxidationsmittel, das keine Peroxo-Verbindung darstellt, ist das Permanganat-Ion, MnO4−. Sie werden sich vor allem mit der Chemie des Wasserstoffperoxids und in geringerem Maße mit der des Permanganats auseinandersetzen. Es sollen dabei die folgenden Fragen untersucht werden: Worauf beruht die hohe Reaktivität? Wie sieht die Redoxchemie von Wasserstoffperoxid aus? Welche qualitativen und quantitativen Methoden gibt es, Wasserstoffperoxid nachzuweisen? Welche Bedeutung besitzt Permangant in der quantitativen analytischen Chemie?
Wissenswertes vorab
Redox-Reaktionen, die Übertragung von Elektronen, sind in der Technik und in der Natur von zentraler Bedeutung. Die Energiegewinnung in der Atmungskette beruht zum Beispiel auf einer kaskadenartig abgestuften Reihe unterschiedlicher Redoxsysteme. Dabei kann es zu einer Fehlleitung von Elektronen kommen. So kann beim Kontakt von Sauerstoff mit 1-Elektronen-Reduktionsmitteln wie NADH oder Ferredoxinen das Hyperoxid-Radikal O2•− entstehen. Hyperoxid (in der Biochemie meist „Superoxid“ genannt) ist eine reaktive Sauerstoff-Spezies, die zellschädigende Eigenschaften besitzt. Die Zelle besitzt daher Enzyme, die Superoxiddismutasen, die das Hyperoxid-Radikal-Anion zu Wasserstoffperoxid (H2O2) und Sauerstoff umwandeln. Das durch Superoxiddismutasen oder durch 2-Elektronen-Reduktionsmittel wie FADH2 erzeugte Wasserstoffperoxid ist ebenfalls zelltoxisch und wird daher durch weitere Enzyme, die Katalasen, in die untoxischen Produkte Sauerstoff und Wasser disproportioniert. Als Beispiel wird Ihnen die Häm-Katalase begegnen, deren aktives Zentrum dem sauerstofftransportierenden Häm-Zentrum im Hämoglobin vom Aufbau her ähnlich ist, aber ein Eisen(III)-Zentralatom in einem Porphyrin-Ringsystem aufweist. Die H2O2-Disproportionierung erfordert einen 2-Elektronen-Redoxprozess. Dabei wird die Eisen(III)-Ruheform des Enzyms um zwei Elektronen oxidert, die entstehende Verbindung kehrt anschließend wieder in die Ruheform zurück (Katalysatoren gehen unverändert aus der Reaktion hervor).
Wasserstoffperoxid kann über katalytisch wirksame Eisen(II)-Ionen in einer als Fenton-Reaktion bezeichneten Redoxreaktion organische Substrate oxidieren. Wasserstoffperoxid wird dabei von Eisen(II) zu Hydroxid und dem stark zelltoxischen Hydroxyl-Radikal reduziert.
Fe2+ + H2O2 → Fe3+(OH−) + HO•
Auch andere Metallionen wie Mn2+ können über eine Fenton-analoge Reaktion reaktive Sauerstoffspezies erzeugen. Sie werden sich daher mit der Redoxchemie von Eisen und Mangan beschäftigen.
Wasserstoffperoxid ist eine schwache Säure und bildet zwei Reihen von Salzen, die Hydroperoxide und die Peroxide. Die in reiner Form blaue Flüssigkeit kommt als 30%ige wässrige Lösung in den Handel („Perhydrol“). Wasserstoffperoxid zersetzt sich unter Wärme- und Lichteinwirkung sowie in Gegenwart katalytischer Mengen an Schwermetallen und Alkalien. Der dabei zunächst in statu nascendi (in der Entstehung) befindliche Sauerstoff ist besonders reaktiv und hat daher eine hohe Bleich- und Desinfektionswirkung. In den Handel kommt Perhydrol deshalb mit Stabilisatoren wie Phosphorsäure.
Wasserstoffperoxid stellt für aerobe Organismen nicht nur ein Zellgift dar. So wird es zum Beispiel kurz nach der Befruchtung von der Eizelle gebildet. Durch Aushärtung der Eiweiße in der Eizellenhülle wird das Eindringen weiterer Spermien verhindert. Auch Pflanzen nutzen Wasserstoffperoxid. Weißfäulepilze bauen den Holzbestandteil Lignin (ein vernetztes Polyphenol) mit Hilfe von Wasserstoffperoxid ab, um an ihr eigentliches Substrat, Cellulose und Hemicellulose, zu gelangen. Das Holz wird dabei weiß und fasrig (daher „Weißfäule“). Das Wasserstoffperoxid wird hierbei von einer Manganperoxidase zu Wasser reduziert, wobei MnII zu MnIII oxidiert wird. Letzteres wird chelatisiert, dringt in das Lignin ein und spaltet dieses oxidativ. In der Technik spielt H2O2 eine wichtige Rolle als Desinfektionsmittel (Abwasseraufbereitung) und Bleichmittel. In seiner Funktion als Bleichmittel findet es Anwendung in der Zellstoffbleiche, wo Restlignin zum Vergilben des Papiers führt. Um Haare zu blondieren oder Zähne zu bleichen wird es oft als an Carbamid gebundenes Peroxid verwendet. Die bleichende Komponente von Waschmitteln ist meist Perborat. Sowohl aus Perboraten wie auch aus Percarbonaten wird H2O2 bei Temperaturen oberhalb 60 °C freigesetzt. In Waschmitteln, die für das Waschen bei niedrigerer Temperatur verwendet werden, sind Bleichaktivatoren enthalten (häufig TAED, Tetraacetylethylendiamin), so dass bereits bei 30 °C eine Bleichwirkung entfaltet wird. Nicht nur Wasserstoffperoxid, sondern auch Peroxo-Verbindungen wie Perborat oder Percarbonat sind gegenüber Schwermetallen empfindlich. So reichen bereits geringe Spuren von Fe, Cu und Mn, um die Reinigungskraft herabzusetzen und wäscheschädigende Verbindungen entstehen zu lassen. Daher werden Bleichstabilisatoren wie edta und Phosphonate eingesetzt, welche die Metallionen binden.
Permanganat ist das Anion der in wässriger Lösung starken und unbeständigen Permangansäure (HMnO4). Das Säureanhydrid (Mn2O7) kann durch vorsichtiges Einwirken von konzentrierter Schwefelsäure auf trockenes Permanganat gewonnen werden (was Sie aber bitte bleiben lassen!). Das in der Durchsicht dunkelrote, in der Aufsicht grünmetallische Öl kann mit überschüssigem kaltem Wasser zu Permangansäure reagieren. Beim Erwärmen zersetzt sich das Anhydrid schlagartig in Braunstein und Sauerstoff. Seine Dämpfe sind aufgrund der Hydrolyse durch Luftfeuchtigkeit violett.
Lernziele und einführende Literatur
Lernziele: Bestimmung von Oxidationszahlen, Aufstellen von Redoxgleichungen, Erkennen von Oxidationsmittel und Reduktionsmittel, Erkennen der pH-Abhängigkeit von Redoxreaktionen, Reaktionen des Wasserstoffperoxids, Disproportionierung und Komproportionierung, Wirkungsweise von Bleichmitteln, Redoxtitration.
Einführende Literatur:
Mor9-229K14.2, Mor9-230K14.3, Mor9-358K21.7
Mor10-241K15.2, Mor10-242K15.3, Mor10-366K22.7
Redox-Reaktionen
Während bei Säure-Base-Reaktionen ein Ungleichgewicht in der Elektronenverteilung ausgeglichen wird, ohne dass Elektronen oder Elektronenpaare vollständig von einem Reaktionspartner auf den anderen übertragen werden, geschieht bei einer Redox-Reaktion genau das. Bei einer Reaktion wie der von Natrium mit Chlor zu Natriumchlorid ist der Elektronenübergang offensichtlich. Bei der Reaktion von Wasserstoff mit Chlor zu Chlorwasserstoff und dessen anschließender Protolyse in Wasser entsteht aber in zwei Schritten auch Chlorid – aber wo genau ist hier der Elektronenübergang?
Oxidation wird als Entzug von Elektronen definiert, Reduktion als Aufnahme von Elektronen. Es werden Elektronen von einem Reaktionspartner auf den anderen übertragen, es findet eine Redox-Reaktion statt. Der Partner, der den anderen oxidiert und dabei selbst reduziert wird, ist das Oxidationsmittel, der Partner, der den anderen reduziert und dabei selbst oxidiert wird, ist das Reduktionsmittel. Man beachte die Verwandtschaft der Konzepte: bei einer Redox-Reaktion werden Elektronen zwischen zwei Partnern ausgetauscht, bei der Protolyse Protonen. In beiden Fällen steht der Austausch im Mittelpunkt, nicht die isolierte Teilreaktion, die nur auf dem Papier formuliert werden kann.
Oxidationszahlen
Um die Elektronenbilanz auch bei einer Reaktion wie der Reduktion von Kupfer(II) durch Glucose aufstellen zu können, ist ein Konzept nötig, mit dessen Hilfe auch nichtionische Stoffe in die Betrachtung einbezogen werden können. Dies wird durch das Konzept der Oxidationszahl geleistet. Regeln zum Ermitteln von Oxidationszahlen können Sie dem Mortimer entnehmen.
Mit Hilfe dieser Regeln ergeben sich die Oxidationszahlen der Atome in vielen Verbindungen und Ionen. Stellen Sie fest, ob Sie ein Problem haben bei: NaCl, MgCl2, SO42−, PO43−, Na3PO4, K2Cr2O7, FeO, Fe2O3, NH3, N2H4, NH2OH, N2, N2O, NO, N2O3, NO2, HNO3.
Nach Einführung der Oxidationszahlen kann alternativ definiert werden: Oxidation ist die Erhöhung der Oxidationszahl, Reduktion deren Erniedrigung. Negativ ausgedrückt: eine Reaktion ohne Oxidationszahländerung ist keine Redox-Reaktion – diese Regel gilt streng und hilft, manchen Irrtum zu vermeiden.
Redoxgleichungen
Sind Ausgangsstoffe und Endprodukte einer Redox-Reaktion bekannt(!), so kann eine Reaktionsgleichung, hier eine Redox-Gleichung, aufgestellt werden. Um die stöchiometrischen Faktoren zu berechnen, wird am Besten für die beiden Redoxpaare getrennt formuliert; anschließend werden die erhaltenen Teilgleichungen unter Berücksichtigung der Elektronenbilanz verknüpft. Hier als Bespiel die Auflösung von Kupfer in halbkonzentrierter Salpetersäure, wobei Kupfer(II)-Ionen und Stickstoffmonoxid entstehen. Da Salpetersäure eine starke Säure ist, wird mit der protolysierten Form H3O+ + NO3− formuliert (dies ist kein kritischer Punkt, wer mag, kann auch von HNO3 ausgehen):
Die Teilgleichung für das konjugierte Redoxpaar 1 ist problemlos aufzustellen:
Cu → Cu2+ + 2 e−
Die Teilgleichung für das konjugierte Redoxpaar 2 wird auf folgende Weise entwickelt:
Das redoxaktive Element: die Oxidationszahlen der beteiligten Atome zeigen, bei welchem Element eine Änderung eingetreten ist (hier bei N):
NVO−II3− → NIIO−II
Die Die Zahl der übertragenen Elektronen: die Differenz der Oxidationszahlen ist die Zahl der Elektronen, welche die reduzierte Form mehr besitzt als die oxidierte Form:
NVO3− + 3 e− → NIIO
Die Ladungsbilanz: die Summe der Ladungen auf jeder Seite der Gleichung muss gleich sein. Auf die Seite mit überschüssiger negativer Ladung werden im Fall einer sauren Lösung H3O+-Ionen zugefügt, im Fall einer basischen Lösung werden OH−-Ionen auf die Seite mit geringerer negativer Ladung zugefügt:
NVO3− + 3 e− + 4 H3O+ → NIIO
Die Stoffbilanz wird ausgeglichen: Die Zahl der Atome jeder Atomsorte muss auf beiden Seiten der Gleichung gleich sein; der Ausgleich erfolgt durch Wasser:
NVO3− + 3 e− + 4 H3O+ → NIIO + 6 H2O
Fertig. Nun kommt noch die Kombination der Teilgleichungen; hierzu werden die beiden Teilgleichungen so mit Faktoren multipliziert, dass die Elektronenzahlen in beiden Teilgleichungen gleich sind (kleinstes gemeinsames Vielfaches der Elektronenzahlen der Teilgleichungen); die Redoxgleichung wird dann durch Addition der beiden Teilgleichungen mit jetzt gleicher Elektronenzahl erhalten:
Cu → Cu2+ + 2 e− × 3
NVO3− + 3 e− + 4 H3O+ → NIIO + 6 H2O × 2
3 Cu + 2 NVO3− + 6 e− + 8 H3O+ → 3 Cu2+ + 6 e− + 2 NIIO + 12 H2O
oder, nach Subtrahieren der auf beiden Seiten auftretenden Teilchen, hier der 6 e−:
3 Cu + 2 NO3− + 8 H3O+ → 3 Cu2+ + 2 NO + 12 H2O
Sie können dem Mortimer zahlreiche weitere Bespiele entnehmen. Mortimer geht dabei etwas anders vor und vermeidet das Aufstellen von Teilgleichungen, um schneller zum Ziel zu gelangen. Sie sollten sich mit beiden Vorgehensweisen vertraut machen, manchmal bietet sich eher die eine Methode an, manchmal die andere. Sehen Sie im Mortimer auch nach, was unter Disproportionierung und Komproportionierung verstanden wird.
Standardpotentiale
Wird eine der beiden Teilgleichungen einer Redoxreaktion als Gleichgewicht betrachtet, so beschreibt das Redoxpotential E seine Lage. Die unter Standardbedingungen gemessenen Redoxpotentiale sind meist nach zunehmenden Werten tabelliert. Die Abfolge wird elektrochemische Spannungsreihe genannt. Die Tabelle erlaubt es zu beurteilen, ob unter Standardbedingungen eine bestimmte Redoxreaktion freiwillig abläuft. Ein hohes Potential bedeutet, dass das Oxidationsvermögen der oxidierten Form hoch ist, dass also die reduzierte Form besonders stabil ist. Einzelpotentiale elektrochemischer Halbzellen können nicht gemessen werden, so wie auch Teilgleichungen keine realen Reaktionen beschreiben. Messbar sind nur Zellspannungen, die Potentialdifferenzen zwischen zwei zusammengeschalteten Halbzellen, die den beiden Teilprozessen einer Redoxreaktion entsprechen. Die tabellierten Standardpotentiale beziehen sich meist auf die Normalwasserstoffelektrode (NWE, auch Standardwasserstoffelektrode) als der einen Halbzelle, deren Potential willkürlich zu null gesetzt wird. Halbzellen mit Standardpotentialen oberhalb von null (zum Beispiel die „edlen“ Metalle) oxidieren die NWE, solche mit Standardpotentialen unterhalb von null (zum Beispiel die „unedlen“ Metalle) reduzieren die NWE unter Wasserstoffentwicklung. (Einige für dieses Projekt relevante Standardpotentiale können Sie dem Anhang des Projektes entnehmen.) Eine Kombination zweier Halbzellen zu einer elektrochemischen Zelle wird als galvanisches Element bezeichnet. Eine Halbzelle mit einem Metall (reduzierte Form), das in eine Lösung eines Salzes desselben Metalls (oxidierte Form) eintaucht, kann direkt in einen Stromkreis eingebunden werden. Die in diesem Projekt oft auftretenden Potentiale nichtmetallischer Halbzellen – wozu auch die Standardwasserstoffelektrode mit ihrer Teilgleichung 2 H3O+ + 2 e− ⇌ H2 + 2 H2O zählt – benötigen ein Hilfsmetall, dass selbst nicht in die Redoxreaktion eingreift (bei der NWE Platin, das sich mit dem unedleren H2 belädt und sich dann wie eine Wasserstoffelektrode verhält).
Die Spannung, die zwischen den Elektroden anliegt, kann aus den Standardpotentialen berechnet werden. Beachten Sie hierbei die Vorzeichenregeln (siehe die oben angegebene einführende Literatur). In diesem Projekt wird es darum gehen, aus der Spannungsreihe den freiwilligen Ablauf einer Redoxreaktion zu ermitteln. Die hierzu verwendete Aussage, dass die reduzierte Form eines Redoxpaares freiwillig mit der oxidierten Form eines darunterstehenden Redoxpaares mit höherem Potential reagiert, ist Ausdruck des Zusammenhangs zwischen der Potentialdifferenz ΔE und der freien Reaktionsenthalpie ΔG. Eine Reaktion läuft freiwillig ab, wenn ΔG < 0. Wegen
ΔG = −n F ΔE
gilt dies für ΔE > 0 (n ist die Zahl der übertragenen Elektronen, F die Faraday-Konstante.
Einen tieferen Einblick in diese Grundregeln erhalten Sie im Projekt „Elektrochemie“.
Vorversuche
In den Vorversuchen werden die typischen Eigenschaften des Kaliumpermanganats und vor allem des Wasserstoffperoxids untersucht.
Nachweisreaktionen für Wasserstoffperoxid
Wasserstoffperoxid kann über zwei gängige Nachweisreaktionen erkannt werden: mit Titanylsulfat als Peroxotitan-Kation oder mit Dichromat als Chromperoxid.
Wasserstoffperoxid reagiert mit Dichromat-Ionen in schwefelsaurer Lösung unter Bildung von tiefblauem Chromperoxid CrO(O2)2. Das Produkt ist in wässriger saurer Lösung instabil, kann jedoch durch geeignete organische Verbindungen, zum Beispiel Ether, einige Zeit stabilisiert werden.
Eine mit verdünnter H2SO4 oder HNO3 angesäuerte K2Cr2O7-Lösung wird mit einigen mL Ether überschichtet und anschließend gekühlt (Eisbad). Dann läßt man 3%-ige Wasserstoffperoxid-Lösung behutsam an der Wand des schräg gehaltenen Reagenzglases einlaufen. An der Phasengrenze zwischen dem Ether und der wässrigen Lösung bildet sich ein tiefblauer Ring. Befinden sich größere Mengen H2O2 in der Probe, so färbt sich die Etherphase vollständig blau. Nach einiger Zeit kann die Farbe in Grün oder Violett umschlagen.
Recherchieren Sie die Strukturformel des Chromperoxids wie sie in der Literatur angegeben wird und erläutern Sie, wozu mit Ether überschichtet wird. (Beachten Sie, dass die lehrbuchübliche Strukturformel bislang nicht experimentell vollständig belegt worden ist.) Erklären Sie an Hand der üblichen Lewisformel und der Reaktionsgleichung, weshalb es sich bei der Umsetzung von Dichromat mit Wasserstoffperoxid zu Chromperoxid nicht um eine Redoxreaktion handelt.
Erkären Sie den nach einiger Zeit stattfindenden Farbumschlag in der wässrigen Phase, für welchen eine Redoxreaktion verantwortlich ist.
Disproportionierung von Hyperoxid
Das Hyperoxid-Radikal ist ein besonders reaktives Teilchen. Es ist in wässriger Lösung instabil und disproportioniert bei der Umsetzung eines Hyperoxids mit Wasser in Sauerstoff und Wasserstoffperoxid, das durch eine charakteristische Orangefärbung nach Zusatz von Titanylsulfat nachgewiesen werden kann. „Titanylsulfat“ ist Titan(IV)-oxid-sulfat, TiOSO4.
1 Spatelspitze Kaliumhyperoxid, KO2, bildet bei der Zugabe zu 25 mL Wasser sofort Sauerstoff und Peroxid. Das entstandene Peroxid wird als Peroxotitanyl-Kation nachgewiesen. Hierzu werden 2 Tropfen der Lösung im Reagenzglas mit 2 Tropfen 3 m H2SO4 angesäuert und mit 1–2 Tropfen 0,1 m Titanylsulfatlösung versetzt. Eine gelbe bis gelborange Färbung, die auf der Bildung von [Ti(O2)(H2O)n]2+ beruht, zeigt H2O2 an.
Formulieren Sie die Gesamt- und Teilgleichungen der Zersetzung von Kaliumhyperoxid. Wann wird eine Redoxreaktion als Disproportionierung bezeichnet?
Durch eine Superoxiddismutase wird diese ohnehin rasche Reaktion um mehrere Größenordnungen beschleunigt, was die Brisanz des Superoxids unterstreicht. Unter Berücksichtigung der Aciditäten (pKS-Werte: HO2 4,8; H2O2 12) kann die katalysierte Reaktion für den physiologischen pH-Wert formuliert werden gemäß:
2 O2•− + 2 H+ → O2 + H2O2
Katalase-katalysierte Zersetzung von Wasserstoffperoxid
Katalasen zersetzen das Zellgift Wasserstoffperoxid zu Wasser und Sauerstoff. Katalasen finden sich in allen aerob lebenden Organismen.
In zwei Reagenzgläser werden 5 mL 3%-ige Wasserstoffperoxidlösung gefüllt. In das eine wird eine sehr geringe Menge an Katalase aus Rinderleber zugegeben, das andere dient als Vergleich.
Achten Sie darauf, die Katalase stets auf Eis aufzubewahren.
Beschreiben Sie ihre Beobachtung. Erklären Sie den Begriff Katalyse allgemein. Skizzieren Sie die Wirkungsweise eines Katalysators am Beispiel der Katalase.
Formulieren Sie für die Redoxreaktion die Gesamt- und Teilgleichungen.
Erklären Sie an Hand der Standardpotentiale, weshalb Wasserstoffperoxid zur Disproportionierung neigt.
Redoxchemie des Wasserstoffperoxids
Im vorangegangenen Versuch wurde die katalysierte Disproportionierung von Wasserstoffperoxid untersucht. Stoffe, die zur Disproportionierung neigen, sich selbst also oxidieren und reduzieren können, wirken anderen Stoffen gegenüber entweder als Reduktions- oder als Oxidationsmittel, je nach dem elektrochemischen Potential des gewählten Stoffes.
Ist ein Redoxpotential pH-abhängig wie zum Beispiel bei CrIII/CrVI, kann H2O2 sogar gegenüber demselben Redoxpaar in Abhängigkeit vom pH-Wert einmal als Oxidations- und einmal als Reduktionsmittel auftreten. Prüfen Sie dies durch Reaktion von H2O2 mit Chrom(III)-Salzlösung und Chromat(VI)-Lösung bei verschiedenen pH-Werten.
Je 5 mL gesättigter Chrom(III)-sulfat-Lösung werden in zwei Reagenzgläser gegeben; je 5 mL gesättigter Kaliumdichromat(VI)-Lösung werden in zwei weitere Reagenzgläser gefüllt. Zu jeder Lösung wird 1 Tropfen 30%-ige H2O2-Lösung zugefügt. In das erste und dritte Glas geben Sie tropfenweise 1 m Schwefelsäure, in das zweite und vierte tropfenweise 2 m Natronlauge. Ermitteln Sie durch pH-Papier den pH-Wert, bei dem sichtbar eine Redoxreaktion stattfindet.
Bei welchem pH-Wert wirkt H2O2 als Reduktionsmittel, bei welchem als Oxidationsmittel? Formulieren Sie die Reaktionsgleichungen und erklären Sie das beobachtete experimentelle Ergebnis.
Weshalb ist das Redoxpotential für das Redoxpaar CrIII/CrVI pH-abhängig?
Ordnen Sie qualitativ die in der Reaktion auftretenden konjugierten Redoxpaare in der richtigen Reihenfolge in einer Redoxreihe an (einmal für die Reaktion im Basischen, einmal für die Reaktion im Sauren).
Reduzierende Wirkung von Wasserstoffperoxid
Wasserstoffperoxid kann durch starke Oxidationsmittel wie Permanganat zu Sauerstoff oxidiert werden. Die Reaktion, die folgendem Handversuch zugrunde liegt, kann zur quantitativen Analyse von Wasserstoffperoxid dienen, welche Ihnen im weiteren Verlauf des Projekts begegnet.
Eine Spatelspitze Kaliumpermanganat wird in 3 m Schwefelsäure gelöst. Anschließend wird 3%-ige H2O2-Lösung zugegeben. Das entstehende Gas wird durch die Glimmspanprobe untersucht.
Stellen Sie die Teil- und Gesamtgleichungen auf. Erklären Sie an Hand der Standardpotentiale und des Zusammenhangs zwischer freier Standardenthalpie ΔG und der Potentialdifferenz ΔE, warum zu erwarten ist, dass die Reaktion freiwillig abläuft.
Oxidierende Wirkung von Wasserstoffperoxid
In der Technik werden Chlor, Chlordioxid, Wasserstoffperoxid und andere Peroxo-Verbindungen sowie Ozon als Bleichmittel eingesetzt. Wasserstoffperoxid gehört dabei zu den Stoffen, die in der „grünen Chemie“ (engl. green chemistry, fr. chimie douce) als ökologisch unbedenkliche Oxidationsmittel eingestuft werden, da es nur Wasser hinterlässt und nicht etwa Salzsäure, Chlorid, oder chlorierte Verbindungen, wie dies bei der Verwendung von Chlor geschieht.
In schwach saurer Lösung oxidert Wasserstoffperoxid Iodid zu Iod. Im folgenden Versuch wird entstandenes Iod durch die Iod-Stärke-Reaktion nachgewiesen.
Ca. 2 mL 3%-ige Wasserstoffperoxid-Lösung werden im Reagenzglas mit verdünnter Essigsäure angesäuert und mit 1 Tropfen 0,05 m Kaliumiodid-Lösung und 1–2 Tropfen Stärkelösung versetzt. In der Kälte tritt sofort Blaufärbung ein. Anschließend wird das Gemisch mit dem Bunsenbrenner bis zur Entfärbung erwärmt und die Lösung danach in ein Eisbad gestellt.
Erklären Sie Färbung und Entfärbung der Probe.
Stellen Sie die Teilgleichungen und die Gesamtgleichung des Redoxprozesses auf. Erklären Sie an Hand der Redoxpotentiale, weshalb die Reaktion von Wasserstoffperoxid und Iod freiwillig abläuft.
Synthese eines stabilisierten Bleichmittels: „Carbamid-Peroxid“
„Carbamid-Peroxid“ ist ein Wasserstoffperoxid-basiertes Bleichmittel, bei dem durch die Bildung einer Additionsverbindung zwischen dem Peroxid und Harnstoff eine stabilere Formulierung vorliegt als bei einer Peroxid-Lösung. Die korrekte Bezeichnung ist Wasserstoffperoxid–Harnstoff (1/1), H2O2·(NH2)2CO.
Eine gesättigte Harnstofflösung wird hergestellt, indem man 5 g Harnstoff in der Kälte in möglichst wenig Wasser löst (Kochsalz/Eis-Kältemischung). Dazu werden 10 mL 30%-ige H2O2-Lösung gegeben. Es fallen farblose, durchscheinende Kristalle des Addukts aus. Stehenlassen im Eisbad erhöht die Ausbeute. Man nutscht die Kristalle ab und wäscht das Produkt zwei- bis dreimal mit einer 1:1-Mischung aus Eiswasser und Methanol, die ca. 0,1%-ig an Citronensäure ist: Wasserstoffperoxid–Harnstoff (1/1) ist unter einem pH-Wert von ca. 4 stabil. Achtung: Da man mit möglichst konzentrierten Lösungen in der Kälte arbeiten muss, tritt das Problem auf, dass anstelle des gewünschten Produkts Harnstoff auskristallisieren kann. Diesen erkennt man aber an den typischen nadelförmigen Kristallen, während Wasserstoffperoxid–Harnstoff (1/1) kompakte Kristallblöcke bildet. Einige Kristalle werden nach dem Trocknen in Wasser gelöst und mit einem geeigneten Nachweis auf Wasserstoffperoxid untersucht.
Betrachten Sie die Struktur von „Carbamidperoxid“. Worin könnte die Stabilisierung des Wasserstoffperoxids in der Additionsverbindung begründet sein?
Auschnitt aus der Kristallstruktur (Cambridge-Datenbank, Eintrag UREXPO11) von Wasserstoffperoxid–Harnstoff (1/1). Rot: Sauerstoff, blau: Stickstoff, grau: Kohlenstoff, kleine Kreise: Wasserstoff.
Darstellung von Kaliumperoxodisulfat durch Elektrolyse von Schwefelsäure
Bei der Elektrolyse von Kaliumhydrogensulfat fällt an der Anode Kaliumperoxodisulfat, K2S2O8, als farbloses Pulver aus. Durch Hydrolyse von Kaliumperoxodisulfat kann Wasserstoffperoxid gewonnen werden. (Der Versuch wird in Zweiergruppen ausgeführt, so dass an jedem der ersten drei Versuchstage mindestens drei Gruppen die Geräte nutzen). Das Produkt wird für die Vollanalyse aufbewahrt!
Eine konzentrierte, saure KHSO4-Lösung wird hergestellt, indem 15 g KHSO4 in 50 mL 1 m Schwefelsäure gelöst werden. Die Elektrolyse wird in einem Becherglas durchgeführt, welches mit einer Kochsalz-Eis-Kältemischung gekühlt wird (die Lösung muss während des gesamten Versuches in einem Temperaturbereich von 0–7 °C gehalten werden). Es wird für mindestens 2,5 Stunden bei ca. 3 A an Platinelektroden elektrolysiert (dabei kommt es zu einer heftigen Gasentwicklung an der Kathode). Nach der Elektrolyse wird die Suspension im Glasfilter abgesaugt und mit eiskaltem Wasser gewaschen. Als Produkt wird eine farblose, feinkristalline Substanz erhalten, von der anschließend möglichst wenig wieder in Wasser gelöst wird. Die Lösung zeigt die typische Nachweisreaktion auf Oxidationsmittel, indem nach Kaliumiodid-Zugabe Iod entsteht.
Worauf bei der Versuchsdurchführung zu achten ist: Bei zu geringer Konzentration an Hydrogensulfat-Ionen würde anstelle des gewünschten Produkts Sauerstoff an der Anode entstehen. Bei zu niedrigen Temperaturen bildet sich an der Anode Ozon.
Anmerkung für die Assistenten: Pro Saal stehen 2-3 Elektroden bereit. Aus Materialgründen muss die Elektrolyse daher in Zweiergruppen durchgeführt werden. An jedem Tag des Projektes "Reaktive Sauerstoffspezies" sollten 2 Gruppen elektrolysieren.
Welches Gas entwickelt sich an der Kathode? Formulieren Sie die Reaktionsgleichung.
Berechnen Sie Ihre Ausbeute, indem Sie mit dem theoretisch zu erwartenden Wert (Faradaysches Gesetz) vergleichen.
Was passiert, wenn Sie verdünnte Schwefelsäure (ohne Kaliumhydrogensulfat) elektrolysieren?
Wasserstoffperoxid wurde früher über die Elektrolyse von höher konzentrierter Schwefelsäure gewonnen. Das Peroxodisulfat-Ion ist in wässriger Lösung beständig; die Säure hingegen wird schnell hydrolysiert. Im ersten Schritt hydrolysiert die Peroxodischwefelsäure (Marshallsche Säure) zu Schwefelsäure und Peroxomonoschwefelsäure (auch Carosche Säure genannt, wässrige Lösungen werden als Piranhasäure bezeichnet), welche in langsamer Reaktion einer weiteren Hydrolyse unterliegt, wobei sich erneut Schwefelsäure bildet, daneben entsteht Wasserstoffperoxid. Formulieren Sie die Teil- und Gesamtgleichungen der Elektrolyse sowie die Hydrolysegleichungen. Ist die Hydrolyse-Reaktion eine Redoxreaktion? Begründen Sie ihre Antwort.
Die Elektrolyse zu Peroxodisulfat ist historisch von Bedeutung, da so Wasserstoffperoxid großtechnisch hergestellt werden konnte. Heutzutage erfolgt die großtechnische Darstellung über das Anthrachinon-Verfahren.
Ähnlich wie das Peroxodisulfat-Ion ist das Molekül des Tetrathionat-Ions, S4O62−, aufgebaut. Anstelle der Peroxo-Gruppierung im S2O82− ist dabei nun eine -S-S-Brücke zu finden. Tetrathionat spielt in der analytischen Chemie eine wichtige Rolle, es bildet sich beispielsweise bei der Oxidation von zwei Thiosulfat-Ionen.
Zeichnen Sie jeweils die Lewis-Formeln für das Thiosulfat- und das Tetrathionat-Ion. Überlegen Sie außerdem, wie die Oxidationstufen festzulegen sind.
Als geeignetes Oxidationsmittel, um Thiosulfat zu Tetrathionat zu oxidieren, kann elementares Iod fungieren. Dieses Prinzip findet in einer speziellen Methode auf dem Gebiet der Redoxtitrationen eine Anwendung, der sogenannten Iodometrie.
Formulieren Sie die Redoxgleichung für die Reaktion von elementarem Iod mit einer Thiosulfatlösung.
Iodometrische Bestimmung von Kupfer
Die Iodometrie basiert auf der oxidierenden Wirkung von elementarem Iod bzw. auf der reduzierenden Wirkung von Iodid. Einerseits können Reduktionsmittel in neutraler Lösung direkt mit einer eingestellten Iod-Maßlösung titriert werden. Andererseits wandeln Oxidationsmittel Iodid in elementares Iod um. Die freigewordene Menge an Iod kann somit mit einer eingestellten Natriumthiosulfat-Maßlösung bestimmt werden. Die Indikation des Auftretens bzw. Verschwindens von elementarem Iod am Äquivalenzpunkt kann durch Zugabe einer Stärkelösung erfolgen. Iod bildet mit löslicher Stärke (Amylose) eine tiefblaue Einschlussverbindung. Diese Farbreaktion ist recht empfindlich, sie ist noch deutlich sichtbar bei Konzentrationen bis zu 10−5 mol L−1. Diese hohe Empfindlichkeit der Iod-Stärke-Reaktion ist allerdings an die gleichzeitige Anwesenheit von Iodid-Ionen gebunden, die mit Iod Polyiodid-Ketten (z. B. I5−) bildet, die sich schließlich in die kanalartigen Hohlräume der schraubenförmig angeordneten Molekülketten der Amylose einlagern.
Kupfer(II)-salze reagieren mit überschüssigem Iodid zu schwerlöslichem Kupfer(I)-iodid bei gleichzeitiger Oxidation von Iodid zu elementarem Iod. Das ausgeschiedene Iod wird im unmittelbaren Anschluss mit einer Thiosulfat-Lösung titriert.
Formulieren Sie die Redoxgleichung für die Reaktion von Cu2+ mit überschüssigen Iodid-Ionen. Informieren Sie sich außerdem in Lehrbüchern, welche anderen Anionen analog reagieren.
Analyse 9 (man.): Kupfer iodometrisch
Zur Bestimmung des Kupfergehaltes werden 50 mL der Analysenlösung mit 50 mL einer 1 m Schwefelsäure versetzt. Anschließend werden 4 g festes Kaliumiodid zugegeben und nach dessen Auflösung die rötlich-braune Lösung mit 0,1 m Natriumthiosulfatlösung titriert. Gegen Ende der Titration, wenn die Lösung nur noch schwach gelb gefärbt ist, fügt man 2 mL Stärkelösung hinzu und titriert zügig bis zum ersten völligen Verschwinden der Blaufärbung.
Mangan: Das chemische Chamäleon
Mangan kommt in seinen Verbindungen in den Oxidationsstufen II bis VII vor. Je nach Oxidationsstufe des Mangans hat die jeweilige Verbindung eine andere Farbe. Auf Grund der Vielfalt der realisierbaren Oxidationsstufen und der damit verbundenen Farben wird Mangan auch als „chemisches Chamäleon“ bezeichnet. Sehr schön lassen sich die verschiedenen Wertigkeitsstufen des Mangans durch Umsetzung von Permangant mit Perborat beobachten.
Zu einigen ml verdünnter Kaliumpermanganat-Lösung (1:10) wird eine kleine Spatelspitze Perborat zugegeben. Es wird mit 3 m NaOH versetzt. Innerhalb von 1–2 Minuten werden verschiedene Farbtöne durchlaufen. Erscheint die Lösung schließlich braun, so wird noch mit 2 m HCl angesäuert.
Halten Sie die Farbtöne in der Reihenfolge ihres zeitlichen Auftretens fest und recherchieren Sie, welche Oxidationsstufe zu welcher Farbe gehört.
Ein Einblick in die Redoxchemie des Mangans
Permanganate sind starke Oxidationsmittel. Sie können daher aus Mangan(II)-Salzen nur mit sehr starken Oxidationsmitteln erzeugt werden. Im folgenden Versuch oxidieren Sie Mn2+ mit Hilfe von PbO2 zu MnO4−.
Die Lösung eines Mangan(II)-Salzes wird in einem Reagenzglas mit etwa demselben Volumen konzentrierter Salpetersäure versetzt und anschließend vorsichtig eine Spatelspitze PbO2 zugegeben. Spätestens nach dem Aufkochen ist eine deutliche Violettfärbung der Lösung zu erkennen.
Stellen Sie die Teil- und Gesamtgleichungen für die Redoxreaktion auf.
Warum kann mit PbO2 Mn2+ zu MnO4− oxidiert werden, wenn doch das Standardpotential des Redoxpaares PbO2/Pb2+ etwas geringer ist als das von MnO4−/Mn2+? Argumentieren Sie an Hand der Reaktionsgleichung.
Woher rührt die intensive Farbe des Permanganat-Ions?
Übungsanalysen
Sowohl für die Übungs- als auch für die Vollanalyse werden Sie sich der Methode der Redoxtitration bedienen. Die Verwendbarkeit von Redoxtitrationen für die Maßanalyse beruht auf der Tatsache, dass bei Redoxvorgängen notwendigerweise die Zahl der abgegebenen Elektronen gleich der Zahl der aufgenommenen Elektronen ist. Das bedeutet, dass zwischen den Stoffmengen an Oxidations- und Reduktionsmittel ein aus der Reaktionsgleichung genau bekannter stöchiometrischer Zusammenhang besteht. Die in ihrer Menge zu bestimmende Verbindung wird also je nach ihren Eigenschaften mit einer Lösung eines Oxidations- oder Reduktionsmittels mit genau bekanntem Gehalt titriert. So wie bei einer Säure-Base-Titration der Äquivalenzpunkt durch einen pH-Sprung gekennzeichnet ist, ändert sich bei einer Redoxtitration im Äquivalenzpunkt sprunghaft das elektrochemische Potential. Die Größe des Potentialsprungs beeinflusst die Titrationsgenauigkeit. Sie ist durch den Unterschied der Standardpotentiale gegeben. Um den Potentialsprung sichtbar zu machen, werden Redoxindikatoren verwendet, falls nicht – wie beim Permanganat – die Eigenfarbe des einen Reaktionspartners ausreicht. Redoxindikatoren sind Farbstoffe, die durch Oxidation oder Reduktion meist reversibel ihre Farbe ändern, also selbst Redoxsysteme darstellen. Das Umschlagsintervall des Indikators muss innerhalb des Bereiches des Potentialsprungs liegen, so wie ein Indikator bei der Neutralisationsanalyse im Bereich des pH-Sprungs umschlagen muss.
Titerstellung einer Kaliumpermanganat-Maßlösung
Die quantitative Analyse von Reduktionsmitteln gelingt durch permanganometrische Titration. Die Permanganometrie ist eine Methode der Redoxanalyse, bei der als Titrant Permanganat-Ionen verwendet werden. Diese sind sehr starke Oxidationsmittel, die im Sauren in Gegenwart von Reduktionsmitteln zu Mangan(II)-Ionen reduziert werden. In neutraler oder schwach alkalischer Lösung erfolgt die Reduktion zum Mangan(IV)-oxid (Braunstein).
Es wird zunächst der Faktor der im Saal vorhandenen 0,02 m Kaliumpermanganat-Lösung bestimmt. Die Faktorbestimmung erfolgt im Sauren mit Oxalat gemäß der folgenden Reaktion.
2 MnO4− + 5 C2O42− + 16 H3O+ → 2 Mn2+ + 10 CO2 + 24 H2O
Einige Gramm Natriumoxalat werden in einem offenen Wägegläschen ca. eine Stunde bei 200–300 °C im Trockenschrank getrocknet (Zersetzung erfolgt erst oberhalb von 330 °C). Nach dem Verschließen des Wägegläschens lässt man im Exsikator erkalten. In mehrere Erlenmeyerkolben werden Mengen zwischen 100–150 mg analysengenau eingewogen, in ca. 70 mL Wasser gelöst und mit 20–30 mL 1 m Schwefelsäure angesäuert. Man lässt die Lösung aufkochen und titriert solange mit der einzustellenden Permangantlösung, bis der erste Tropfen mindestens 30 Sekunden lang nicht mehr entfärbt wird. Eventuell können zu Beginn einige Tropfen MnSO4-Lösung zugefügt werden.
Anmerkung: Verwenden Sie gemeinsam mit ihrem Labornachbarn eine Flasche mit KMnO4-Maßlösung. Bestimmen Sie beide den Faktor und notieren Sie diesen auf der Flasche. Benutzen Sie für sämtliche ihrer Titrationen die Maßlösung dieser Flasche, so dass Sie den Faktor nicht ständig neu bestimmen müssen.
Beachten Sie beim Arbeiten mit Kaliumpermanganat-Lösungen, dass deren Zersetzung durch katalytisch wirksame Staubkörnchen oder Glasoberflächen beschleunigt wird. Da Licht solche Reaktionen weiter beschleunigt, werden Kaliumpermanganat-Lösungen in dunklen Glasflaschen aufbewahrt. Da die unerwünschten Reaktionen dadurch aber nur verlangsamt werden, muss von Zeit zu Zeit der Titer der Lösung neu bestimmt werden.
Wie eine wässrige H2O2-Lösung, so ist auch eine wässrige Kaliumpermanganat-Lösung thermodynamisch instabil. An Hand der Potentiale müsste jeweils welche Zerfallsreaktion eigentlich ablaufen? Verwenden Sie die im Anhang angegebenen Redox-Potentiale, um die Zerfallsreaktionen zu erkennen; formulieren Sie die Zerfallsgleichungen. Die beiden wässrigen Lösungen werden in der Literatur als metastabil bezeichnet, was ist damit gemeint?
Warum müssen bei der Titerstellung, die in der Hitze erfolgt, örtliche Permangant-Überschüsse vermieden werden?
Vollanalyse
Neben Perboraten und anderen Peroxo-Verbindungen finden Chlor, Chlordioxid und Ozon Anwendung als Bleich- und/oder Desinfektionsmittel. Zur Desinfektion von Wasser, zum Beispiel in Schwimmbädern, wird oft Chlor eingesetzt – entweder als Gas oder durch die Reaktion von Salzsäure und Natriumhypochlorit. In der Vollanalyse bestimmen Sie den Gehalt einer peroxidhaltigen Lösung. Die hier ausgegebenen Lösungen können Ihnen im Alltag beim Haare blondieren, Wäsche waschen und bei der Schimmelpilzbekämpfung begegnen.
Bestimmung einer peroxidhaltigen Probe
Analyse 10 (man.): Peroxid-Bestimmung
Die ausgegebene peroxidhaltige Probe wird mit ca. 100 mL Wasser verdünnt und mit 30 mL 1m Schwefelsäure angesäuert. Es wird mit Wasser bis zur 250-mL-Marke aufgefüllt. Dann werden 25 mL der entstandenen Mischung mit 25 mL Schwefelsäure angesäuert und mit Wasser auf 100 mL verdünnt. Es wird solange mit der Permanganatlösung titriert, bis die erste Rosafärbung mindestens 30 Sekunden bestehen bleibt.
Stellen Sie die Gleichung für die Reaktion zwischen Wasserstoffperoxid und Permanganat auf. Berechnen Sie die Menge an Wasserstoffperoxid im 250-mL-Messkolben in mg.
Bei der folgenden Analyse am Titrierautomaten sollte die erhaltene Titrationskurve etwa so aussehen:
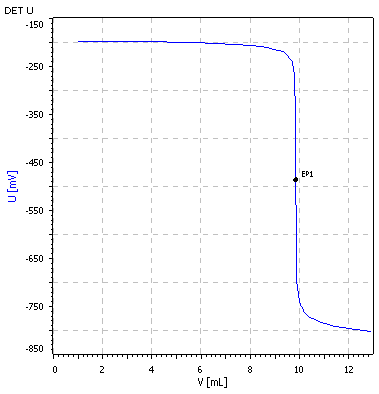
@ Analyse 10 (aut.): Peroxid-Bestimmung (Platinelektrode)
Führen Sie die gleiche Titration, die Sie von Hand ausgeführt haben, am Titrierautomaten durch. Verwenden Sie hierzu möglichst kleine Rührfische, um Verwirbelungen und Gasblasen an der Elektrode zu vermeiden.
Übungsanalysen
Permanganometrische Bestimmung von Nitrit
Nitrit neigt unter bestimmten Bedingungen genauso wie Wasserstoffperoxid zur Disproportionierung. Seine Darstellung kann aus NO und NO2 erfolgen, welche in Alkalilaugen zu Nitrit komproportionieren (formulieren Sie die Reaktionsgleichung).
Analyse 11 (man.): Nitrit-Bestimmung
Der Messkolben mit der Analysenlösung wird bis zur 250-mL-Marke aufgefüllt. Anschließend wird die Bürette mit dieser Lösung befüllt. In einen Erlenmeyerkolben werden 25 mL 0,02 m KMnO4-Maßlösung vorgelegt, mit Wasser auf 100 mL verdünnt, mit 40 mL 1 m Schwefelsäure angesäuert und auf 40 °C erwärmt. Die Bürettenspitze wird in die Lösung eingetaucht. Dann wird langsam bis zur gerade eintretenden Entfärbung titriert, wobei mit einem Magnetrührer intensiv gerührt wird. Besonders gegen Ende der Titration sollten nicht mehr als drei Tropfen pro Minute zugesetzt werden. (Kann man im Verlauf der Titration auch nur Spuren von nitrosen Gasen an der Kolbenöffnung riechen, hat man zu schnell titriert und wird wahrscheinlich ein zu niedriges Ergebnis finden.) Bei sehr langer Dauer der Titration sollte die Temperatur kontrolliert und eventuell wieder auf 40 °C eingestellt werden.
Stellen Sie die Gleichung für die Reaktion zwischen Nitrit und Permanganat auf. Berechnen Sie die Menge an Nitrit im 250-mL-Messkolben in mg.
Formulieren Sie die Reaktionsgleichung für die Komproportionierung von NO mit NO2 indem Sie Teil- und Gesamtgleichungen aufstellen.
Warum müssen Sie zur Bestimmung von Nitrit mittels Permanganometrie einen Umweg (umgekehrte Titration) einschlagen? Warum tauchen Sie die Bürettenspitze in die saure Lösung?
Bestimmung von FeII manganometrisch
Analyse 12 (man.): Eisen manganometrisch
Die ausgegebene eisenhaltige Probe wird mit ca. 100 mL Wasser verdünnt und mit 30 mL 1m Salzsäure angesäuert. Es wird mit Wasser bis zur 250 mL-Marke aufgefüllt.
50 mL der Analysenlösung werden mit 20 mL Reinhardt-Zimmermann-Lösung versetzt und mit Wasser auf ca. 100 mL verdünnt. Es wird solange mit 0,02m Kaliumpermanganatlösung titriert, bis die erste Rosafärbung mindestens 30 Sekunden bestehen bleibt.
Berechnen Sie die Menge an FeII im 250-mL-Messkolben in mg.
Vollanalyse
Eine Peroxoverbindung, die Sie bereits in der Elektrolyse kennen gelernt haben, ist das Peroxodisulfat. Peroxodisulfate werden erst bei Temperaturen oberhalb von 60 °C durch Sauerstoff-Abspaltung als Bleichmittel wirksam.
Bestimmung von Kaliumperoxodisulfat
Da Kaliumperoxodisulfat nicht direkt mit Permanganat bestimmt werden kann, läßt man die Probe zunächst auf eine bekannte, überschüssige Menge Eisen(II)-sulfat einwirken, wobei dieses zu Eisen(III)-sulfat oxidiert wird. Anschließend wird der Teil an Eisen(II)-sulfat, der nicht reagiert hat, mit Permanganat titriert. Parallel wird die gleiche Menge Eisen(II)-sulfat ohne Probenzusatz titriert. Über die Differenz der benötigten Volumina an Permanganat-Maßlösung kann auf die unbekannte Menge an Peroxodisulfat geschlossen werden.
@ Analyse 13 (man.): Peroxodisulfat-Bestimmung
Zur Bestimmung werden 1,2–2,1 g des aus der Elektrolyse gewonnenen Peroxodisulfats analysengenau eingewogen und in einem 250-mL-Messkolben gelöst. Für die Eisen(II)-Lösung werden in einem 250-mL-Messkolben 8 g „Mohrsches Salz“, (NH4)2Fe(SO4)2·6H2O, in Wasser gelöst, mit 30 mL 1 m Schwefelsäure angesäuert und mit Wasser auf 250 mL aufgefüllt. (Die Lösung ist ca. 0,08-molar an Eisen.)
Titration 1: 25 mL Eisenlösung werden mit 25 mL 1 m Schwefelsäure angesäuert und mit Wasser auf 100 mL verdünnt. Anschließend wird mit Permanganat solange titriert, bis gerade ein Farbumschlag von leicht grünlich nach rosa auftritt. Man erhält Verbrauch 1.
Titration 2: 25 mL Peroxodisulfat-haltige Probenlösung werden mit 25 mL Eisenlösung versetzt und mit 25 mL 1 m Schwefelsäure angesäuert. Mit Wasser wird auf 100 mL verdünnt. Anschließend wird mit Permanganat solange titriert, bis gerade ein Farbumschlag von leicht grünlich nach schwach orange auftritt. Man erhält Verbrauch 2, der zwischen einigen Tropfen und Verbrauch 1 liegen sollte. Verwenden Sie für alle Schritte ausgekochtes Wasser.
(1) Stellen Sie sämtliche für die Bestimmung relevanten Reaktionsgleichungen auf. (2) Berechnen Sie die Menge an Peroxodisulfat im 250-mL-Messkolben in mg. (3) Geben Sie aus der Abweichung von ihrer eingewogenen Menge an Peroxodisulfat die Reinheit ihres Elektrolyseproduktes in Prozent an.
Geben Sie mögliche Gründe für die Unreinheit Ihres Elektrolyseproduktes an.
Bestimmung von Kaliumperoxodisulfat mittels Titrierautomat
@ Analyse 13 (aut.): Peroxodisulfat-Bestimmung (Platinelektrode)
Führen Sie die gleichen Titrationen, die Sie von Hand ausgeführt haben, am Titrierautomaten durch. Setzen Sie dazu pro Saal 1 L Mohrsches-Salz-Lösung für alle Studierenden an (siehe oben), und benutzen Sie diesselbe Pipette zur Dosierung.
Vergleichen Sie ihre Ergebnisse und erläutern Sie eventuelle Abweichungen.
Diese allgemeinen Konzepte sollten Sie hinter den Versuchen erkennen …
Wasserstoffperoxid ist metastabil und neigt zur Disproportionierung. Es findet als Oxidationsmittel breite Verwendung, es kann aber auch reduzierend wirken. Im Organismus kann es durch partielle Reduktion von Sauerstoff entstehen. Da es ein Zellgift ist, wird es mit Hilfe von Katalasen zu Sauerstoff und Wasser umgesetzt. In der Technik findet es Anwendung als Bleichmittel, oft in Form von Perborat oder Percarbonat. Die quantitative Analyse erfolgt mittels Permanganometrie, der qualitative Nachweis beruht auf der Bildung des Peroxotitan-Kations oder des Chromperoxids. Aufgrund des hohen elektrochemischen Potentials des Redoxpaares Mn2+/MnO4− erlaubt die Permanganometrie die quantitative Bestimmung verhältnismäßig starker Oxidationsmittel.
Welche Verbindungen bilden die Alkalimetalle beim Verbrennen an der Luft? Wie reagieren diese mit Wasser?
Warum dürfen hypochlorithaltige Haushaltsreiniger nicht gemeinsam mit salzsäurehaltigen Reinigungsmitteln verwendet werden? Stellen Sie die zur Beantwortung der Frage nötige Reaktionsgleichung auf und erläutern Sie, um welche Art von Reaktion es sich hierbei handelt.
Wasserstoffperoxid wird im Allgemeinen als Oxidationsmittel verwendet. Stellen Sie die Reaktionsgleichungen für die Reaktionen von Wasserstoffperoxid mit (a) salpetriger Säure und (b) schwefliger Säure in saurer Lösung und (c) mit Chrom(III) in alkalischer Lösung auf.
Gegenüber stärkeren Oxidationsmitteln wirkt Wasserstoffperoxid reduzierend. Formulieren Sie die Reaktion von H2O2 mit (a) Permanganat, (b) Bleidioxid, und (c) Ozon. Bestimmen Sie die Oxidationszahlen.
Der pKS-Wert von Wasserstoffperoxid beträgt 12. Welches Mengenverhältnis von H2O2 und HO2− enthält eine Waschflotte vom pH-Wert 11?
Was versteht man unter Rücktitration, indirekter und umgekehrter Titration?
… noch ein paar Tipps, wie Sie Fehler vermeiden können:
Peroxid-Bestimmung: (1) Führen Sie die Analyse so zügig wie möglich durch; lassen sie Lösungen nicht unnötig lange herumstehen. (2) Da beim Befüllen des Meßkolbens ein Schäumen auftreten kann, warten Sie für das Auffüllen bis zur Eichmarke bis die Schaumbildung aufgehört hat.
Für das Projekt sind zwei Wochen vorgesehen. Achten Sie daher bei Ihrer Zeitplanung darauf, die erste Woche mit der Peroxidbestimmung am Titrierautomaten abzuschließen.
Anhang: Standardpotentiale in saurer Lösung (pH = 0)
Entnommen aus: P. W. Atkins, Physikalische Chemie, 3. Auflage, Wiley-VCH, Weinheim, 2001.
| ox. | red. | n e− | E0/V |
|---|---|---|---|
| Sn2+ | Sn | 2 | 0,14 |
| Hg2Cl2 | 2 Hg + 2 Cl− | 2 | 0,27 |
| I2 | 2 I− | 2 | 0,54 |
| O2 + 2 H3O+ | H2O2 + 2 H2O | 2 | 0,70 |
| Fe3+ | Fe2+ | 1 | 0,77 |
| Hg2+ | Hg | 2 | 0,86 |
| 2 Hg2+ | Hg22+ | 2 | 0.92 |
| MnO2 + 4 H3O+ | Mn2+ + 6 H2O | 2 | 1,23 |
| O2 + 4 H3O+ | 6 H2O | 4 | 1,23 |
| Cr2O72− + 14 H3O+ | 2 Cr3+ + 21 H2O | 6 | 1,33 |
| PbO2 + 4 H3O+ | Pb2+ + 6 H2O | 2 | 1,46 |
| Mn3+ | Mn2+ | 1 | 1,51 |
| MnO4− + 8 H3O+ | Mn2+ + 12 H2O | 5 | 1,51 |
| 2 HOCl + 2 H3O+ | Cl2 + 4 H2O | 2 | 1,63 |
| H2O2 + 2 H3O+ | 4 H2O | 2 | 1,78 |
| S2O82− | 2 SO42− | 2 | 2,05 |
Farbe
Warum ist ein grünes Leuchten in zahlreichen Fernsehkriminalfällen ein Indiz für ein Verbrechen? Warum brennen Lithium-Salze mit roter Flamme und warum erscheint eine Suspension von Goldnanopartikeln blau? Woher kommen diese Farbeindrücke? Farbreaktionen und -eindrücke bestimmen das tägliche Leben und sind auch ein wesentlicher Bestandteil der Analytischen Chemie. Tatsächlich haben „Farben“ ganz verschiedenen Ursachen. In diesem Projekt werden grundlegende Phänomene, die zu Farbeindrücken führen, unterschieden und Messmethoden vorgestellt. Die erarbeiteten physiko-chemischen Kenntnisse bilden die Grundlage für sämtliche weiteren Praktika in den verschiedenen Lehrbereichen.
Wissenswertes vorab
Licht: Besteht aus elektromagnetischen Wellen, die Energie mit bestimmter Wellenlänge und Ausbreitungsrichtung transportieren.
Absorption: Absorption von sichtbarem Licht führt zu einer elektronischen Anregung der Materie, die Intensität des einfallenden Lichts wird dadurch abgeschwächt.
Fluoreszenz: Nach Absorption von Licht können manche Materialien die aufgenommene Energie in Form von sichtbarem Licht, als Fluoreszenz oder Phosphoreszenz, wieder abgeben.
Streuung: Elastische Lichtstreuung führt nur zu einer Änderung der Ausbreitungsrichtung.
Optische Spektroskopie: Absorption, Fluoreszenz und Streuung einer Probe sind stoffspezifische Eigenschaften. Die spektral aufgelöste Bestimmung dieser Eigenschaften durch die optische Spektroskopie ermöglicht somit die zerstörungsfreie Analyse von Stoffen.
Farbreaktionen: Chemische Reaktion, bei deren Verlauf eine Farbänderung auftritt.
Lernziele und einführende Literatur
Lernziele: Ursache von Farbeindrücken, Licht als elektromagnetische Welle und Licht als Photonen, Lichtintensität als Funktion der Wellenlänge, Spektrum, Absorption, Streuung, Fluoreszenz, Extinktion, Spektrometer, UV/Vis-Spektroskopie, Lambert-Beer-Gesetz, Flammenfärbung, dispersive Elemente, Polarisator, optische Filter, additive/subtraktive Farbmischung, Polarisation, Kontinuum, Linienspektrum.
Einführende Literatur: Vorlesungsskript, Atkins
Vorversuche
Spektrale Zerlegung einer Weiß-Lichtquelle
Weißes Licht besteht aus verschiedenen Spektralanteilen, die mittels eines dispersiven Elements räumlich aufgespalten werden können. Dispersive Elemente sind beispielsweise Prismen oder optische Beugungsgitter. Auch feine Wassertropfen können Licht in seine spektralen Bestandteile aufteilen und so einen Regenbogen verursachen.
Im ersten Vorversuch sollen die Funktionsweisen verschiedener dispersiver Elemente untersucht werden.
Untersuchen Sie die optischen Eigenschaften eines Prismas, eines Beugungsgitters und eines Handspektroskops mit Hilfe verschiedener Lichtquellen (z. B. Neonröhre, Sonne, Taschenlampe). Betrachten Sie dazu das ausfallende bzw. reflektierte Licht unter verschiedenen Winkeln. Variieren Sie auch den Einfallswinkel. Betrachten sie des Weiteren die Reflexion einer Taschenlampe auf der Unterseite einer CD und einer DVD.
Skizzieren sie den Strahlengang des Lichts durch das Prisma und die Aufspaltung des Lichts. [2P]
Aus welchen sichtbaren Spektralanteilen besteht das Licht der verschiedenen Lichtquellen? [2P]
Was lässt sich bei der Beleuchtung einer CD-Unterseite/DVD-Unterseite mit der Taschenlampe beobachten? [1P]
Welche Unterschiede zwischen der CD und der DVD lassen sich erkennen und was ist die Ursache hierfür? [2P]
Spektrale Analyse verschiedener Lichtquellen
Der Vorversuch dient dem Verständnis der Eigenschaften und Unterschiede zwischen einem Linienspektrum und einem Kontinuum.
Führen Sie eine spektrale Analyse des Lichts einer Neonröhre, einer Taschenlampe und einer LED durch. Verwenden Sie hierzu ein Faserspektrometer und ein Handspektroskop.
Verwenden Sie dazu das Spektrometer im Scope-Modus
![]() mit einer Integrationszeit von 3,8 ms, 10 Scans zur Mittelwertbildung und einer Boxcar Breite von 2.
mit einer Integrationszeit von 3,8 ms, 10 Scans zur Mittelwertbildung und einer Boxcar Breite von 2.
Achten Sie darauf, dass die Ausleuchtung des Spektrometers 65000 Counts nicht übersteigt! Richten Sie deshalb das offene Ende der Faser nie direkt in die Lichtquelle, sondern nähern diese vorsichtig von der Seite an.
Stellen Sie die Spektren der verschiedenen Lichtquellen in Graphen dar. [2P]
Wodurch unterscheiden sich die Spektren der Lichtquellen?
Wann erscheint eine Lichtquelle „weiß“, wann mit einer „bestimmten Farbe“? [2P]
Polarisation
Das Licht einer natürlichen Quelle ist unpolarisiert. Polarisatoren dienen zur Selektion bestimmter Polarisationsebenen und können somit auch als Analysatoren verwendet werden. Optisch aktive Substanzen wie beispielsweise D(+)-Glucose können die Polarisationsrichtung des Lichtes drehen. Diese Eigenschaft kann als Nachweis für die Reinheit von optisch aktiven Substanzen verwendet werden.
Betrachten Sie eine Lichtquelle durch zwei Folienpolarisatoren während Sie einen der beiden drehen.
Überprüfen Sie die Formel aus der Vorlesung für die Transmission als Funktion des Winkels zwischen den beiden Polarisatoren qualitativ. [2P]
Betrachten Sie das Licht einer Deckenlampe durch einen Folienpolarisator nachdem es an einer glatten Oberfläche reflektiert wurde. Versuchen Sie dabei eine Auslöschung des reflektierten Lichts zu beobachten.
Begründen Sie Ihre Beobachtungen im Fall des reflektierten Lichts. Was kann hieraus über die Lichtausbreitung gefolgert werden? [2P]
Wie kann aus diesem Versuch die Transmissionsrichtung des Analysators bestimmt werden? [1P]
Extinktion, Absorption und Transmission verschiedener Farbfilter und farbiger Lösungen
Es werden die Effekte von RGB und CMY Farbfiltern untersucht. Dabei wird der Zusammenhang zwischen Farbeindruck und Spektrum betrachtet. Die Wirkung von farbigen Lösungen als Farbfilter wird anhand eines pH-Indikators bei verschiedenen pH Werten betrachtet.
Die Begriffe Extinktion, Absorption und Transmission werden eingeführt und unterschieden.
Messen Sie qualitativ die Absorptionsspektren aller 6 Farbfilter mit Hilfe des UV/Vis-Spektrometers. Verwenden Sie dazu das Spektrometer im Scope-Modus ![]() mit einer Integrationszeit von 3,8 ms, 10 Scans zur Mittelwertbildung und einer Boxcar Breite von 2.
mit einer Integrationszeit von 3,8 ms, 10 Scans zur Mittelwertbildung und einer Boxcar Breite von 2.
Achten Sie darauf, dass die Ausleuchtung des Spektrometers 65000 Counts nicht übersteigt! Richten Sie deshalb das offene Ende der Faser nie direkt in die Lichtquelle, sondern nähern diese vorsichtig von der Seite an.
Verwenden Sie als Lichtquelle eine Taschenlampe und nehmen Sie davon zunächst ein Spektrum auf. Messen Sie nun für jeden Farbfilter jeweils ein Spektrum, indem Sie den Farbfilter zwischen Taschenlampe und dem offenen Ende der Faser einbringen.
Stellen Sie die Spektren der RGB Filter zusammen mit dem der Taschenlampe in einem Graphen dar. Erstellen Sie einen zweiten Graphen entsprechend mit den Spektren der CMY Filter und der Taschenlampe. [2P]
Wie entsteht der Farbeindruck nach einem Farbfilter? Erläutern Sie den Unterschied zwischen den RGB und den CMY Filtern jeweils anhand eines Beispiels. [2P]
Zur Aufnahme von Absorptionsspektren verschiedener Indikatorlösungen verfahren Sie wie im Abschnitt Aufnahme von Absorptionsspektren erläutert ist. Verwenden Sie jeweils eine basische und eine saure Lösung von Methylorange (je 3 mL einer 0,1 m HCl-Lösung und einer 0,1 m NaOH-Lösung
mit je einem Tropfen Indikator versetzt).
Stellen Sie die beiden Absorptionsspektren in einem Graphen dar. Wo liegen die Absorptionsmaxima für Methylorange im Basischen und im Sauren? [1P]
Berechnen Sie anhand der beiden Absorptionsspektren die Transmissionsspektren der Indikatorlösungen. [2P]
Stellen Sie die Reaktionsgleichung für die Reaktion in saurer und basischer Umgebung für Methylorange auf. [1P]
Fluoreszenz
Der Zusammenhang zwischen der Konzentration einer Fluoreszenzfarbstofflösung und der Intensität des Emissionsmaximums soll unter Verwendung eines Fluoreszenzspektrometers untersucht werden.
Betrachten Sie zunächst eine konzentrierte Lösung von Fluorescein aus dem Indikatorsatz.
Welchen Farbeindruck erhalten Sie bei der Betrachtung der konzentrierten Fluoresceinlösung? [1P]
Gegeben ist eine Ausgangslösung von Fluorescein in Ethanol bekannter Konzentration. Stellen Sie eine Verdünnungsreihe mit den Konzentrationen 0,1 mmol L−1, 0,2 mmol L−1, 0,3 mmol L−1, 0,4 mmol L−1, 0,6 mmol L−1, 0,8 mmol L−1 und 1,0 mmol L−1 her und messen Sie die Fluoreszenzemissionsspektren.
Stellen Sie die gemessenen Fluoreszenzspektren in einem Graphen dar. Entnehmen Sie aus den Spektren die Wellenlänge des Emissionsmaximums. Was wäre eine geeignete Anregungswellenlänge? [3P]
Entnehmen Sie an der Wellenlänge des zuvor ermittelten Emissionsmaximums die Intensität bei verschiedenen Konzentrationen. Erstellen Sie daraus ein Diagramm, indem Sie die Intensität gegen die Konzentration auftragen. Bei welchen Konzentrationen befindet man sich bezüglich der Fluoreszenzintensität im linearen Bereich? Woraus resultieren Abweichungen vom linearen Verhalten? [3P]
Übungsanalysen
Anwendung des Lambert-Beerschen Gesetzes
Mit Hilfe des Lambert-Beerschen Gesetzes wird der Extinktionskoeffizient eines Farbstoffs aus der gemessenen Optischen Dichte bei verschiedenen Konzentrationen ermittelt.
Gegeben ist eine 1,0 mmol L−1 Stammlösung von Chinolingelb in Wasser.
Stellen Sie eine Verdünnungsreihe mit den Konzentrationen 0,005 mmol L−1, 0,01 mmol L−1, 0,02 mmol L−1, 0,03 mmol L−1, 0,04 mmol L−1 her. Messen Sie von allen Verdünnungen jeweils ein Absorptionsspektrum.
Stellen Sie die gemessenen Spektren in einem Graphen dar und bestimmen sie die Wellenlänge des Absorptionsmaximums. [2P]
Fertigen Sie ein Diagramm an, indem Sie die Optische Dichte an der Wellenlänge des zuvor bestimmten Absorptionsmaximums gegen die Konzentration auftragen. Passen Sie eine Ursprungsgerade an die Datenpunkte an. Ermitteln Sie den Extinktionskoeffizienten von Chinolingelb mit Hilfe des Lambert-Beerschen Gesetzes aus der Steigung der Regressionsgeraden (Einheit beachten!). [3P]
Warum wird für die Anpassung eine Ursprungsgerade verwendet? [1P]
Warum werden so niedrige Konzentrationen für den Versuch gewählt? [1P]
Flammenfärbung verschiedener Alkali-Metall-Salze
Alle Elemente emittieren im atomaren oder ionisierten gasförmigen Zustand bei hohen Temperaturen für das Element charakteristisches Licht. Bei den Alkali-, Erdalkali- und einigen anderen Elementen lässt sich dies bereits in einer Bunsenbrennerflamme erreichen.
Gegeben sind fünf Proben bekannter Alkali- und Erdalkalisalze. Betrachten Sie die Flammenfärbung der verschiedenen Substanzen. Verwenden Sie dazu ausgeglühte Magnesiastäbchen, mit denen Sie eine kleine Menge der zu untersuchenden Substanz aufnehmen und in die Flamme halten.
Zur Analyse erhalten Sie eine unbekannte Probe und sollen diese einem der fünf bekannten Kationen zuordnen.
Vergleichen Sie die Farbeindrücke der jeweiligen Kationen. [1]
Welches Kation war in der unbekannten Substanz enthalten? [1P]
Warum soll bei der Handhabung der Magnesiastäbchen Hautkontakt vermieden werden? [1P]
Vollanalyse
Konzentrationsbestimmung von Farbstofflösungsgemischen
Unter Verwendung des Lambert-Beerschen-Gesetzes sollen die Konzentrationen der Komponenten eines Lösungsgemisches aus Chinolingelb und Patentblau V ermittelt werden. Der Extinktionskoeffizient für Chinolingelb ist aus der Übungsanalyse bekannt.
Stellen Sie eine Verdünnungsreihe für Patentblau V mit folgenden Konzentrationen her: 0,002 mmol L−1, 0,003 mmol L−1, 0,004 mmol L−1 und 0,005 mmol L−1. Das Lösungsmittel ist Wasser. Messen Sie die Absorptionsspektren.
Bestimmen Sie den Extinktionskoeffizienten für den Patentblau V Farbstoff wie in der Übungsanalyse. [3P]
Messen Sie nun das Absorptionsspektrum des Farbstoffgemisches.
Stellen Sie das Spektrum des Farbstoffgemisches in einem Graphen dar. [1P]
Bestimmen Sie die Konzentrationen von Chinolingelb und Patentblau V mit Hilfe des Labert-Beer-Gesetztes. Warum kann die Bestimmung über die beiden Maxima getrennt behandelt werden? [2P]
Führen Sie eine Fehlerabschätzung durch. Überlegen Sie dazu wo mögliche Fehlerquellen liegen und wie groß die jeweils resultierenden Ungenauigkeiten sind. [2P]
Chemolumineszenz – Luminol – Blutspurentest
Bei der Chemolumineszenz wird bei einer exothermen Reaktion die frei werdende Energie in Form von Licht, meist im sichtbaren Bereich zwischen 400 und 700 nm, ausgestrahlt. Da es sich um keine Temperaturstrahlung handelt, wird es oft als „kaltes Licht“ bezeichnet. Die Chemolumineszenz tritt bei vielen chemischen Reaktionen auf, in denen energiereiche, angeregte (instabile) Zwischenstufen entstehen und sofort wieder, unter Emission von Licht, zerfallen.
Die bekannteste Chemolumineszenz-Reaktion ist die Oxidation von Luminol mit Wasserstoffperoxid in Gegenwart von Eisen- oder Mangan-Ionen, die zur Sichtbarmachung von Blutspuren genutzt wird (der Blutfarbstoff Hämoglobin enthält Eisen-Ionen). Das oxidierte Luminol-Molekül dient dabei gleichzeitig als Sensibilisator. Ein weiteres Beispiel sind kommerziell erhältliche Kunststoffröhrchen, die beim Knicken ein intensives, verschiedenfarbiges, lang anhaltendes Licht abgeben. Dieses Licht wird ebenfalls durch Chemolumineszenz erzeugt. In den Röhrchen befinden sich drei Komponenten: ein Oxalsäureester, ein Farbstoff, dessen Emission die Farbe des Lichtes bestimmt, und ein Röhrchen mit Wasserstoffperoxid. Wird das Röhrchen mit Wasserstoffperoxid zerbrochen, startet die Peroxyoxalat-Chemolumineszenz.
Dieser Versuch findet als Saalversuch statt. Lösung I ist eine basische Luminollösung, Lösung II ist K3[Fe(CN)6] gelöst in Wasserstoffperoxid. Bitte notieren Sie ihre Beobachtungen.
Was beobachten Sie nach Zusammengeben der Lösungen I und II? [1P]
Welche Funktion hat das Kaliumhexacyanoferrat? [1P]
Formulieren Sie die Luminolreaktion. [1P]
Diese allgemeinen Konzepte sollten Sie hinter den Versuchen erkennen
Farbeindrücke können sehr verschiedene Ursachen haben. Hierzu gehören die Absorption, Reflexion und Streuung von Licht, die Aussendung von Licht als Fluoreszenz oder Phosphoreszenz sowie Chemolumineszenz und die hier nicht besprochene Wärmestrahlung.
Weißes Licht resultiert aus der Überlagerung von Lichtwellen unterschiedlicher Farben (Wellenlängen). Farbeindrücke entstehen wenn nur bestimmte Farben vorhanden sind oder wenn Farben „fehlen“. Die Charakterisierung einer Lichtquelle und ihres Farbeindrucks erfolgt anhand ihres Spektrums, welches den spektral abhängigen Intensitätsverlauf aufzeigt.
Sortieren Sie die Farben Rot, Blau, Gelb und Grün nach aufsteigender Wellenlänge. [1P]
Ist natürliches Licht polarisiert? [1P]
Welcher Zusammenhang besteht zwischen der Absorptions- und der Fluoreszenzwellenlänge eines Stoffes? [1P]
Reaktionskinetik
Die Geschwindigkeit mit der eine Reaktion abläuft, kann von Reaktion zu Reaktion stark variieren. So laufen die Explosion von Nitroglycerin oder die Neutralisation von NaOH und HCl innerhalb von Bruchteilen einer Sekunde ab. Die Faltung eines Proteins kann dagegen abhängig von der Größe einige Sekunden bis Minuten dauern, das Rosten von Eisen benötigt schon einige Jahre und die Umwandlung von Diamant zu Graphit verläuft nicht messbar langsam (findet jedoch statt). Bei vielen Reaktionen ist es jedoch wichtig eine quantitative Aussage zur Reaktionsgeschwindigkeit machen zu können. So ist es zum Beispiel in der Pharmakologie wichtig die Wirkungsdauer eines Medikamentes, in der Biologie die Dauer einer enzymatischen Reaktion oder den radioaktiven Zerfall von natürlich vorkommenden Isotopen bei der Altersbestimmung von Gegenständen (zum Beispiel der Zerfall von 14C bei der Radiocarbonmethode) zu bestimmen. Die Reaktionskinetik beschreibt mit mathematischen Ausdrücken die Konzentrationsänderung der Reaktanden und der Produkte in Abhängigkeit von der Zeit und somit mit welcher Geschwindigkeit eine Reaktion abläuft.
Wissenswertes vorab
Betrachtet wird eine Reaktion der Form
A → B
[A] und [B] bezeichnen die Konzentrationen der Reaktanden A und B zu einem beliebigen Zeitpunkt t.
Reaktionsgeschwindigkeit: Die Reaktionsgeschwindigkeit v kann als Bildungsgeschwindigkeit des Produkts B
vB = d[B]/dt
oder als Verbrauchsgeschwindigkeit des Edukts A
vA = -d[A]/dt
angegeben werden.
Geschwindigkeitsgesetz: Die Reaktionsgeschwindigkeit einer Reaktion ist gewöhnlich proportional zu irgendeiner Potenz der Konzentrationen der Reaktanden. Für eine Beispielreaktion etwa von drei Reaktanden A, B und C könnte gelten:
v = k·[A]x·[B]y·[C]z
Die Proportionalitätskonstante k wird als Geschwindigkeitskonstante bezeichnet. Sie ist nicht abhängig von der Konzentration der Reaktanden, hängt aber von der Temperatur T ab. x, y und z geben die Reaktionsordnung an. Die Gesamtreaktionsordnung ergibt sich aus deren Summe.
Das Geschwindigkeitsgesetz ist mathematisch betrachtet eine Differentialgleichung. Durch Integration des Geschwindigkeitsgesetzes wird das integrierte Zeitgesetz erhalten. Aus dem Zeitgesetz der Reaktion und der Lösung der Differentialgleichung, kann die Konzentration eines beliebigen Reaktanden zu einem beliebigen Zeitpunkt vorhergesagt werden.
Reaktionsordnung: Die gesamte Reaktionsordnung entspricht der Summe der Potenzen, mit der die jeweiligen Reaktanden im Geschwindigkeitsgesetz erscheinen. Die Reaktionsordnung muss nicht ganzzahlig sein. Wichtige Spezialfälle sind jedoch Reaktionen 0., 1. und 2. Ordnung.
Übersicht über die einfachsten Reaktionsordnungen
| 0. Ordnung | 1. Ordnung | 2. Ordnung | |
|---|---|---|---|
| Typ | A + Katalysator → B + C + Katalysator | A → B + C | 2A → C + D |
| Geschwindigkeitsgesetz | -d[A]/dt = k0 | -d[A]/dt = k1·[A] | -d[A]/dt = k2·[A]2 |
| integriertes Zeitgesetz | [A] = -k0·t + [A]0 | [A] = [A]0·exp(-k1·t) | 1/[A] = k2·t + 1/[A]0 |
| Halbwertszeiten | (t½)0 = 1/(2·k0)·[A]0 | (t½)1 = ln(2)/k1 | (t½)2 = 1/k2·1/[A]0 |
| Beispielreaktion | von Proteinen katalysierte Reaktionen in einer Zelle | Radioaktiver Zerfall | H2 + I2 → 2 HI oder die Dimerisierung von Butadien in der Gasphase |
ACHTUNG:
Aus der Reaktionsgleichung kann nicht direkt auf die Reaktionsordnung geschlossen werden!
Betrachtet wird die Reaktion:
H2 + Br2 → 2 HBr
Diese ist nicht zwangsweise 2. Ordnung, obwohl dies auf dem Papier zunächst so erscheinen mag!
Reaktionen dieses Typs können zweiter Ordnung sein und es fällt uns leicht dies mit der Reaktionsgleichung in Einklang zu bringen. Wir stellen uns vor, die Edukte A und B stoßen zusammen und reagieren zu den Produkten C (und ggf. D).
Die Reaktionsordnung hängt allerdings vom Reaktionsmechanismus einer Reaktion ab. Dieser ist aus der Reaktionsgleichung nicht immer ersichtlich. Die obenstehende Reaktion verfügt über eine einfache Stöchiometrie, allerdings über einen komplizierten Reaktionsmechanismus. So kann es etwa sein, dass nicht die Edukte selbst miteinander reagieren, sondern erst reaktive Zwischenprodukte aus den Edukten entstehen müssen, die dann miteinander zu den Produkten reagieren.
Deshalb kann für die obenstehende Reaktion bei manchen Versuchsbedingungen überhaupt keine Gesamtreaktionsordnung angegeben werden.
In der Praxis muss durch Versuchsreihen der Reaktionsmechanismus aufgeklärt und anschließend auf die Reaktionsordnung zurückgeschlossen werden.
Lernziele und einführende Literatur
Lernziele: Beschreibung der Reaktionskinetik durch mathematische Formeln, Erkennen der Reaktionsordnung durch graphische Auswertung der Messergebnisse
Einführende Literatur: Charles E. Mortimer Chemie, 9. Auflage 2007 (Kapitel 15), Peter W. Atkins, Julio de Paula Physikalische Chemie, 4. Auflage 2006 (Kapitel 22), Peter W. Atkins, Julio de Paula Kurzlehrbuch Physikalische Chemie, 4. Auflage 2008 (Kapitel 15)
Vorversuche
Beobachtung von unterschiedlich schnell ablaufenden Reaktionen.
Reaktion von Kristallviolett
Kristallviolett ist ein organischer Farbstoff aus der Klasse der Triphenylmethyl-Farbstoffe. Das Molekül kann bei Bestrahlung
Licht einer
bestimmten Wellenlänge aus dem sichtbaren Bereich des elektromagnetischen Spektrums absorbieren.
Wir nehmen schließlich die Komplementärfarbe des
absorbierten Lichts als Farbeindruck wahr. Im Fall des Kristallvioletts bedeutet das: Die Substanz absorbiert das gelbe Licht
und sie erscheint deshalb violett.
Die Farbigkeit des Moleküls basiert auf einem über das ganze Molekül delokalisierten π-Elektronensystem. Durch die
Delokalisierung wird das Absorptionsmaximum des Farbstoffs in den sichtbaren Bereich des elektromagnetischen Spektrums
verschoben. Die Delokalisierung findet jedoch nur statt,
wenn das Molekül eine planare Geometrie, ähnlich einer Scheibe, aufweist. Die planare Struktur und das delokalisierte System
gehen verloren, wenn eine Base, z.B. KOH, am zentralen Kohlenstoffatom nukleophil angreift und sich so eine Hydroxygruppe (-OH)
anlagert. Das Molekül absorbiert nun andere Wellenlängen aus dem nicht sichtbaren Bereich des Lichtspektrums und die Substanz
erscheint farblos.
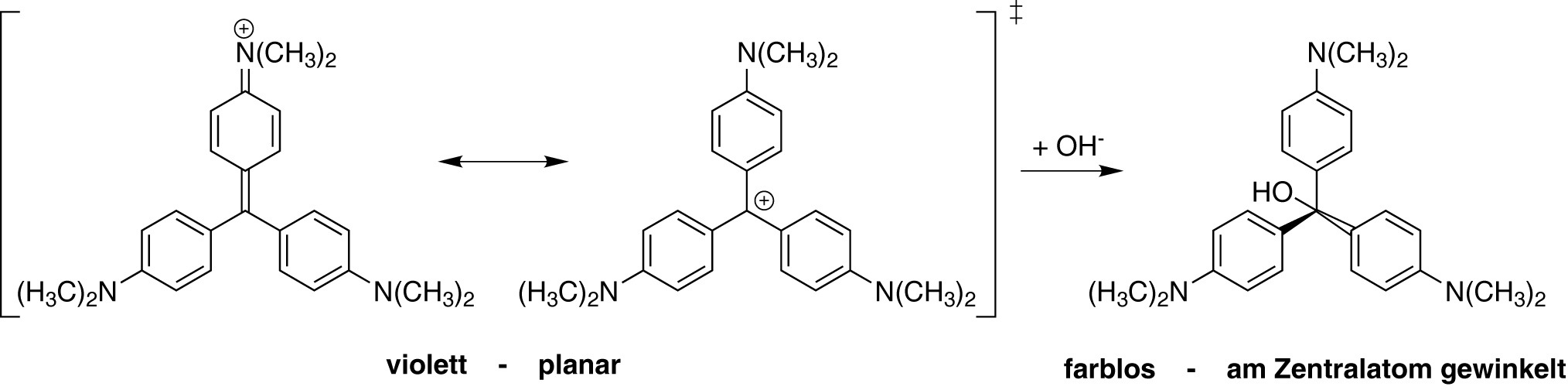
In einem Uhrglas werden 1 Tropfen Kristallviolettlösung aus dem W-Satz und 4 mL Wasser miteinander vermischt. In einem
zweiten Uhrglas werden 1 mL Kristallviolett mit 4 mL 3 m KOH gegeben.
Beschreiben Sie Ihre Beobachtung. [1P]
In welchem Wellenlängenbereich des Spektrums liegt das Absorptionsmaximum vor und nach der Anlagerung des Hydroxidions?
(Wellenlängenbereich angeben!) [2P]
Wieso kann die Reaktion auch mit Carbonaten in wässriger Lösung durchgeführt werden? (Reaktionsgleichung angeben!) [2P]
Wie ist das Zentralatom vor und nach der Anlagerung der Base hybridisiert? Wie groß sind jeweils die Bindungswinkel am Zentralatom? [2P]
Zersetzung von Wasserstoffperoxid
Das Enzym Katalase findet sich in vielen Lebewesen, auch beim Menschen. Es katalysiert die Zersetzung von Wasserstoffperoxid (H2O2). Die Zersetzung von H2O2 ist für Organismen wichtig, da das reaktive H2O2 das Erbgut der DNA oder Proteine schädigen kann. Bei der Zersetzung entsteht auch ein Gas.
Für den Vorversuch benötigen Sie eine handelsübliche Kartoffel. Zerstampfen Sie die Kartoffel in einem Mörser und überführen Sie sie in ein Becherglas. Geben Sie vorsichtig 30%ige
Wasserstoffperoxid-Lösung hinzu.
Beschreiben Sie die Beobachtung. Wie können Sie sie erklären? [2P]
Wie können Sie das entstehende Gas identifizieren? Formulieren Sie die Reaktionsgleichung (Redoxreaktion!) der Zersetzung von Wasserstoffperoxid. [3P]
Beschreiben Sie Ihre Beobachtung. Wie lässt sie sich erklären? [2P]
Vollanalyse Fluoreszenz
Kinetik der Lumineszenz von Leuchtsternen
Leuchtsterne in Kinderzimmern leuchten noch einige Zeit, nachdem das Licht gelöscht wurde. Der folgende Versuch beschäftigt sich mit der Chemie, die hinter den Leuchtsternen steckt. Die Leuchtsterne bestehen aus Zinksulfid, ZnS. ZnS ist namensgebend für den Zinkblende-Strukturtyp. Die Sulfid-Anionen (S2−) bilden eine kubisch flächenzentrierte Kugelpackung (fcc), die kleineren Zn2+ Kationen besetzen die Hälfte der Tetraederlücken (1/2 TL). Außerdem ist Cu1+ in sehr kleinen Konzentrationen eingelagert. Die Oxidationsstufe 1+ für Kupfer ist sehr selten. Für gewöhnlich bevorzugt Kupfer die stabilere Oxidationsstufe 2+. Zwischen den besetzten Orbitalen des ZnS, die das Valenzband (VB) bilden und den unbesetzten (dem Leitungsband, LB) befindet sich eine Bandlücke, in der keine erlaubten Zustände für das ZnS vorhanden sind. Die Orbitale der eingelagerten Kupferionen befinden sich hier nun genau in der Bandlücke des ZnS, sie besetzen die lokalisierten Zustände A. Durch Bestrahlung mit Licht kann ein Elektron aus dem besetzten Valenzband in das unbesetzte Leitungsband angehoben werden (erstes Bild). Das angeregte Elektron hinterlässt so ein positiv geladenes Loch im Valenzband, da nun eine Elektronenladung zu wenig im Valenzband vorhanden ist (zweites Bild). Dieses Ladungsdefizit kann nun durch ein Elektron eines Cu1+ kompensiert werden, das ein Elektron an das Valenzband des ZnS abgibt und somit selbst zu Cu2+ oxidiert wird (zweites und drittes Bild).
Die Lebenszeit des Elektrons im angeregten Zustand liegt typischerweise im ns-Bereich (= 10−9 s), d.h. die angeregten Elektronen werden innerhalb weniger ns wieder ins Valenzband zurückfallen. Dennoch kann durch permanentes Einstrahlen eine gewisse Anzahl von Löchern im Valenzband im zeitlichen Mittel gehalten werden, es wird nach einigen Sekunden eine Sättigung erreicht.
Nun wird die Bestrahlung gestoppt. Es befindet sich eine bestimmte Anzahl von Elektronen im Leitungsband und exakt die gleiche Anzahl an Löchern im Valenzband, da jedes angeregte Elektron genau ein Loch im Valenzband produziert hat. Die Reaktion des Cu1+ verläuft relativ langsam, da sie thermisch aktiviert ist. Dennoch kann sie bei einer ausreichend großen Anzahl von Löchern im VB stattfinden. Somit füllen die Kupferionen mit ihren Elektronen die Löcher im Valenzband auf. Wenn eines der angeregten Elektronen im Leitungsband nun relaxiert, kann es nicht mehr in das Valenzband des ZnS zurückfallen, da das Loch bereits durch ein Elektron eines Kupfers aufgefüllt wurde. Folglich fällt das Elektron in einen Zustand des neugebildeten Cu2+ zurück und reduziert dieses wieder zu Cu1+. Die überschüssige Energie wird beim Zurückfallen als Strahlung abgegeben (drittes Bild). Da die Orbitale des Kupfers energetisch oberhalb der des ZnS-Valenzbandes liegen, verkleinert sich die frei werdende Energie und damit vergrößert sich die Wellenlänge der emittierten Strahlung. Wäre das Elektron in das Valenzband zurückgefallen, wäre Strahlung im UV-Bereich des EM-Spektrums emittiert worden, die für uns nicht sichtbar ist. Die nun emittierte Strahlung liegt im sichtbaren Bereich und wird als grün-gelbe Lumineszenz wahrgenommen. Hier wird die Verweildauer des Elektrons im angeregten Zustand um einige Größenordnungen verlängert, da das angeregte Elektron ein Cu2+ benötigt, um relaxieren zu können. Da die Konzentration der Cu1+ Ionen sehr gering ist, ist die Lumineszenz noch einige Minuten nach Löschen des Lichts beobachtbar (in Dunkelheit sogar einige Tage).
Beachten Sie den Unterschied zum Versuch mit Kristallviolett: Der organische Farbstoff absorbiert Licht einer
bestimmten Wellenlänge des sichtbaren Bereichs (VIS-Bereich). Der Farbstoff „subtrahiert“ mit anderen Worten also eine
Wellenlänge aus dem Weißlicht,
wir nehmen die jeweilige Komplementärfarbe der absorbierten Wellenlänge wahr.
Hier wird hingegen von den Leuchtsternen Licht einer wohldefinierten Wellenlänge des VIS-Bereichs emittiert, d.h. ausgesandt.
Wir nehmen direkt die Wellenlänge des emittierten Lichts wahr.
Zur Durchführung des Versuchs wird eine spezielle Küvette verwendet. In einer Aussparung der Küvette ist Kupfer dotiertes Zinksulfid eingebracht.
Die Küvette wird in den Probenhalter des Spektrometers gestellt. Es soll die
emittierte Strahlung gemessen werden, daher muss der Detektor im 90°-Winkel aufgestellt werden.
Es wird eine Messung vorbereitet, wie dies im Abschnitt Aufnahme von Emissionsspektren beschrieben wird.
Zunächst muss das Fluoreszenzmaximum der Substanz bestimmt werden. Hierfür wird ein Fluoreszenzspektrum aufgenommen. Zur Aufnahme des Fluoreszenzspektrums wird der Haken bei Strobe/Lamp Enable entfernt und die Messung durch Klicken auf Play gestartet. Durch Setzen des Hakens bei Strobe/Lamp Enable wird nun die Probe für einige Sekunden mit Licht bestrahlt. Anschließend wird der Haken bei Strobe/Lamp Enable wieder entfernt. Jetzt kann das Nachleuchten der Substanz durch Klicken auf die Zoomwerkzeuge gut beobachtet werden.
Im Anschluss wird ein Trenddiagramm im Bereich +/- 5 nm um das Fluoreszenzmaximum vorbereitet, aber noch nicht gestartet (siehe dazu den Abschnitt Aufnahme eines Trenddiagramms). Die Messung wird durch Klicken auf den oberen Start-Button gestartet. Durch Setzen des Hakens bei Strobe/Lamp Enable wird die Probe ca. 5 Sekunden mit Licht bestrahlt. Der Haken bei Strobe/Lamp Enable wird wieder entfernt und sehr schnell durch Klicken auf den unteren (kleineren) Start-Button die Messung des Trenddiagramms gestartet. Es wird solange gemessen, bis die Fluoreszenz auf das Level des Hintergrundrauschens abgefallen ist. Zu Beginn des Versuchs sollte idealerweise mindestens eine Intensität von ~ 200 Counts gemessen werden, sodass ein Abfall der Intensität klar beobachtbar ist.
Notieren Sie die im Experiment verwendete Integrationszeit.
Die Auswertung wird mit dem Programm QtiPlot durchgeführt. Dazu müssen die Messdaten exportiert und in QtiPlot eingelesen werden. Der Header der Ausgabedatei wird entfernt. Die erste Spalte beinhaltet lediglich die Uhrzeit der Messung und wird gelöscht. Die beiden verbliebenen Spalten beinhalten die Zeit in Sekunden, die nach Start der Messung vergangen sind und den jeweils zugeordneten Messwert.
Stellen Sie das Fluoreszenzspektrum des Zinksulfids in einem Schaubild dar. [2P]
Rechnen Sie die detektierte Intensität des Trenddiagramms (nicht die des Fluoreszenzspektrums!) in Intensitätswerte pro Sekunde um. Hierzu benötigen Sie die im Experiment verwendete Integrationszeit.
Beim Experiment werden Counts detektiert. Diese sollen in eine Anzahl von Photonen umgerechnet werden, da Photonen im Gegensatz zu den Counts eine anschauliche Bedeutung
als sichtbare Lichtquanten haben. Dem Datenblatt des Detektors wurde entnommen, dass 1 Count 60 Photonen entsprechen. [2P]
Verwenden Sie im weiteren Verlauf der Auswertung stets die Intensitätswerte des Trenddiagramms in Photonen/s
Werten Sie die Versuchsergebnisse des Trenddiagramms graphisch aus. Verwenden Sie die in der obigen Frage berechneten Werte für die Intensität in Photonen/s.
Welche Reaktionsordnung liegt bezüglich der Abklingreaktion vor? (welche Methode wenden Sie an, um die Reaktionsordnung zu bestimmen?)
Fitten Sie die Messdaten an und bestimmen Sie die Geschwindigkeitskonstante k der Abklingreaktion. Welche Einheit hat k? [4P]
Wie lässt sich die gefundene Reaktionsordnung mit dem im obenstehenden Schema erklärten Reaktionsmechanismus in Einklang bringen? [2P]
Das menschliche Auge nimmt in einem dunklen Raum eine Intensität von 60 Photonen/s noch als konstantes Leuchten wahr. Ein gelegentliches Aufleuchten der Substanz kann noch bis
zu einer Intensität von 5 Photonen/s wahrgenommen werden. Berechnen Sie, wie lange Sie das Nachleuchten des Zinksulfids theoretisch in einem abgedunkelten Raum als konstantes
Leuchten bzw. als gelegentliches Aufleuchten beobachten können.
Benutzen Sie hierfür Ihre Messdaten und das integrierte Zeitgesetz für die von Ihnen zuvor bestimmte Reaktionsordnung. [4P]
Wenn Sie den Versuch mit einer Lichtquelle wiederholen würden, die ausschließlich rotes Licht emittiert: Welchen Ausgang des Experiments erwarten Sie und wie lässt sich die Beobachtung erklären? [2P]
Wie sind Fluoreszenz und Phosphoreszenz definiert? Würden Sie nach den von Ihnen gefundenen Definitionen das Nachleuchten des
Zinksulfids als Fluoreszenz oder Phosphoreszenz bezeichnen? [2P]
Vollanalyse Kristallviolett
Wenn sich eine Studentin oder ein Student mit dem Füller verschreibt, greift sie oder er meist zu einem Tintenkiller. Im nachfolgenden Versuch soll verstanden werden, welche Chemie hinter einem Tintenkiller steckt. Tinte besteht unter anderem aus einem organischen Farbstoff, meist einem Triphenylmethyl-Farbstoff. Der Tintenkiller enthält reduzierende Inhaltsstoffe wie Sulfite, Carbonate oder Thiosulfate, die das zentrale Kohlenstoffatom des Farbstoffes reduzieren. Dadurch wird die planare Geometrie zerstört und das farbgebende π-System geht verloren. Folglich entfernt ein Tintenkiller die Tinte nicht, sondern entfärbt den Farbstoff nur. Im folgenden Versuch soll Kristallviolett (Bestandteil von violetter Tinte) durch Zugabe von Base entfärbt werden. Der Reaktionsverlauf wird photometrisch verfolgt.
Kristallviolett
Das Prisma in einem Photometer zerlegt das Licht aus einer Lichtquelle in seine einzelnen Spektralanteile und der Monochromator des Photometers filtert eine Wellenlänge aus dem Spektrum heraus mit der die Probelösung bestrahlt wird. Das Photometer misst die Absorption (das Maß für das Absorptionsvermögen eines Stoffes), indem es die Intensität des hindurchtretenden Lichts einer gefärbten Probelösung I mit der Intensität einer ungefärbten Lösung Io vergleicht:
E = lg (Io/I)
Nach dem Lambert-Beer'schen Gesetz ist die Extinktion einer Lösung proportional zur Schichtdicke der Küvette, der Konzentration der Lösung und dem stoffspezifischen Extinktionskoeffizient. Die Schichtdicke ist mit 1 cm genormt und kann somit in der Gleichung vernachlässigt werden. Für die Referenzlösung gilt ebenfalls das Lambert-Beer'sche Gesetz und durch Gleichsetzen der Gleichungen für beide Lösungen kann die Konzentration der Probelösung berechnet werden:
c = (E/Eo) · co.
Die Reaktion der Entfärbung hängt prinzipiell von den Eduktkonzentrationen ab, die Base liegt jedoch im deutlichen Überschuss vor ([OH−] >> [Kristallviolett]), so dass sich die Konzentration an Lauge vernachlässigbar wenig verändert im Laufe der Reaktion.
Verwenden Sie für den Versuch die im Saal bereitstehende Kristallviolettlösung und nicht die Indikatorlösung!
Notieren Sie sich die Konzentration der Kristallviolettlösung.
Bereiten Sie eine Absorptionsmessung vor, indem sie wie im Abschnitt Aufnahme von Absorptionsspektren beschrieben vorgehen und dabei eine 0,1 m KOH-Lösung gefüllte
Küvette als Referenz verwenden.
Für die Messung des zeitlichen Laufs der Reaktion sollte vor der Entfärbung durch KOH der Absorptionswert des Trenddiagramms zwischen 0,5 und 1,0 OD liegen.
Testen Sie dazu Verdünnungen von 1:1 (das bedeutet 1 Teil Kristallviolett und 1 Teile Wasser), 1:10 und 1:20.
Legen Sie 1 mL der verdünnten Kristallviolettlösung in der Küvette vor und geben 2 mL Wasser hinzu. Messen Sie das Absorptionsspektrum und lesen die Position des Absorptionsmaximums ab. Notieren Sie die verwendete Verdünnung und geben Sie sie in der Auswertung an!
Starten Sie ein Trend-Diagramm, füllen die Menüs entsprechend aus und geben dabei den Bereich von +/− 5 nm um das zuvor bestimmte Absorptionsmaximum an.
Stellen Sie eine Küvette mit 1 mL verdünnter Kristallviolettlösung in den Küvettenhalter und geben sie mit der Mikropipette rasch 2 mL 0,1 m KOH in die Küvette und mischen die Reaktionsmischung durch, indem Sie den Pipettenkolben vorsichtig auf- und abpipettieren.
Klicken Sie auf den oberen Play-Button und setzen Sie einen Haken bei Strobe/Lamp enable.
Durch Klicken auf den unteren Play-Button starten Sie rasch das Trenddiagramm. Nehmen Sie 10 Minuten lang Messdaten auf, bis die Lösung nahezu vollständig entfärbt ist.
Zur Auswertung werden die Messdaten exportiert und in QtiPlot eingelesen. Der Header wird entfernt. Die erste Spalte beinhaltet lediglich die absolute Uhrzeit der Messung und wird gelöscht. Die beiden verbliebenen Spalten beinhalten die Zeit in Sekunden, die nach Start der Messung vergangen sind und den jeweils zugeordneten Absorptionswert. Der Absorptionswert wird nach erfolgreicher Überprüfung der Reaktionsordnung über der Zeit geplottet. Die Daten werden angefittet.
Berechnen Sie die Geschwindigkeitskonstante k' der Reaktion (welche Einheit hat k'?).
Welche Ordnung hat die beobachtete Kinetik? Mit welcher Methode können Sie dies herausfinden? [5P]
Welche Reaktionsordnung ist nach dem Reaktionsmechanismus für die Entfärbung von Kristallviolett zu erwarten? Wie ist dies mit dem Ergebnis Ihres Versuches in Einklang zu bringen? [3P]
Welche möglichen Fehlerquellen spielen bei der Bestimmung von k' eine Rolle? [4P]
Wieso muss für ein genaues Versuchsergebnis eine mit KOH-Lösung gefüllte Küvette als Referenzprobe gemessen werden? [1P]
Hier wird die Absorption gemessen. Der Detektor steht im Strahlengang direkt hinter der Probe. Wird die Fluoreszenz eines Farbstoffes
gemessen, wird der Detektor im 90° Winkel aufgestellt. Wie erklärt sich der Unterschied? [2P]
Vollanalyse Landolt
Der Chemiker Hans Heinrich Landolt untersuchte die zeitlich verzögerte Reaktion von Iodsäure und schweflige Säure, die Landolt-Reaktion. Oft wird sie bei Schauversuchen zur Herstellung von synthetischem „Bier“ genutzt. An diesem Versuch kann anschaulich der Einfluss der Reaktionsparameter auf die Reaktionsgeschwindigkeit und damit die Inkubationszeit beobachtet werden. Als Reaktionsparameter werden die Reaktionstemperatur und die Konzentrationen der Reaktanden untersucht.
Temperaturerhöhung führt zu einer Beschleunigung der Reaktionsgeschwindigkeit. Die Arrhenius-Gleichung beschreibt die Temperaturabhängigkeit der Geschwindigkeitskonstante k. Sie setzt k mit der Temperatur T, der Boltzmannkonstante kb und der Aktivierungsenergie Ea in Beziehung.
k = A · exp(− Ea / kbT).
Damit zwei Teilchen miteinander reagieren können, müssen sie zusammenstoßen. Der präexponentielle Faktor A wird auch Frequenzfaktor genannt und gibt an, wie häufig die Teilchen in einem Zeitintervall zusammenstoßen. Der Faktor Ea im Exponenten beschreibt, welche Energie bei einem Zusammenstoß nötig ist, damit es zu einer Reaktion kommen kann. Anschaulich wird er durch den „Berg“ auf der Reaktionskoordinate zwischen Eduktzustand und Produktzustand beschrieben (siehe unten stehendes Schaubild). Durch eine Erhöhung der Temperatur stoßen die Reaktionspartner häufiger und heftiger gegeneinander und der Anteil der Moleküle mit ausreichender Energie zum Überwinden der Reaktionsbarriere steigt an.
Konzentrations- und Temperaturabhängigkeit der Reaktionsgeschwindigkeit der Landolt-Reaktion
In einer wässrigen Lösung von Iodat und Sulfit ist die ablaufende Reaktion abhängig von dem Verhältnis der Eduktkonzentrationen. Iodat wird von überschüssigem Sulfit zu Iodid reduziert (Reaktion I), bei einem Überschuss an Iodat läuft die Reaktion bis zum Iod ab (Reaktion II). Iodid und Iodat reagieren in saurer Lösung in einer Komproportionierung ebenfalls zu Iod (Reaktion III). Das entstehende Iod kann durch Zugabe von Stärkelösung und Bildung des tiefblauen Iod-Stärke-Komplexes nachgewiesen werden. Bei stöchiometrischem Einsatz der Edukte für Reaktion I setzt die Blaufärbung zeitversetzt ein (Inkubationszeit), bei einem Verhältnis der Eduktkonzentrationen, das die Reaktion II bevorteilt (d.h. bei einem Überschuss an Iodat) ist dagegen eine sofortige Blaufärbung sichtbar. Die zeitverzögerte Reaktion entsteht auf Grund einer ablaufenden Konkurrenzreaktion, in der Iod durch Sulfit schneller reduziert wird (Reaktion IV) als durch Reaktion I und III gebildet werden kann. Deswegen tritt Iod erst in der Lösung auf, wenn alle SO32−-Ionen verbraucht sind. Die Inkubationszeit wurde das erste Mal systematisch von Landolt untersucht.
Hergestellt werden müssen folgende Lösungen: 0,01 m KIO3-Lösung, 0,1
m H2SO4-Lösung, 0,02 m Na2SO3-Lösung
und eine frische Stärkelösung aus 2 g löslicher Stärke und 500 mL Wasser (kurz aufkochen!). Um die Landolt-Reaktion in den
vier unterschiedlichen Verdünnungsstufen bei Raumtemperatur durchzuführen, werden am besten in einem 500 mL Becherglas die Lösungen in den
angegebenen Volumina (siehe Tabelle) bis auf die Na2SO3-Lösung vorgelegt. Die Sulfitlösung wird anschließend
rasch hinzugegeben. Durch kräftiges Umschwenken wird für eine gute Durchmischung der Lösung gesorgt. Vom Augenblick
der Zugabe bis zum Farbumschlag wird die Zeit möglichst genau gemessen.
Um die Inkubationszeit bei unterschiedlicherTemperatur (10 °C und 40 °C) mit gleichen Volumina zu vergleichen, wird die gewünschte Temperatur durch Eintauchen der Lösungen in ein Bad mit heißem Wasser oder mit Eiswasser eingestellt, während des Versuches konstant gehalten und anschließend notiert. Am bsten werden die Volumina verwendet, bei denen bis zum Farbumschlag 10–30 Sekunden vergangen sind.
| KIO3/ mL | Stärke/ mL | Na2SO3/ mL | H2SO4/ mL | Wasser/ mL | Vges/ mL | T/ K | Inkubationszeit/ s |
|---|---|---|---|---|---|---|---|
| 20 | 10 | 20 | 30 | 40 | 120 | ||
| 20 | 10 | 20 | 60 | 130 | 240 | ||
| 20 | 10 | 20 | 90 | 220 | 360 | ||
| 20 | 10 | 20 | 120 | 310 | 480 |
Formulieren Sie die Reaktionsgleichungen der vier einzelnen Reaktionsschritte. [4P]
Erstellen Sie eine Tabelle mit den Inkubationszeiten für die vier Verdünnungsstufen und die verschiedenen Reaktionstemperaturen. Erklären Sie die Änderung der Inkubationszeiten. [2P]
Vollanalyse Katalase
Bestimmung der Reaktionsordnung
Im Vorversuch zur Zersetzung des Wasserstoffperoxids haben Sie qualitativ untersucht, welches Gas bei der Zersetzung frei wird und die Reaktionsgleichung aufgestellt. Mittels des vorliegenden Versuchsaufbaus kann die Kinetik der Zersetzungsreaktion nun quantitativ studiert werden.
In diesem Zusammenhang soll auch der Einfluss der Temperatur auf die Geschwindigkeit der enzymatischen Reaktion getestet werden. Hierzu wird die Reaktion bei verschiedenen Temperaturen durchgeführt.
Im untenstehenden Schaubild ist die Funktionsweise eines Katalysators veranschaulicht. Ein Katalysator setzt die Aktivierungsenergie herab und beschleunigt so die Reaktion. Beachten Sie, dass auf der Abszisse der Reaktionspfad und nicht die Zeit aufgetragen ist. Der Katalysator greift in den Reaktionsmechanismus ein und eröffnet einen neuen, energetisch günstigeren Reaktionspfad auf der die Reaktion ablaufen kann.
Der Versuchsaufbau besteht aus einem Dreihalskolben mit Tropftrichter, Thermometer und einem Schlauch zur Gasabführung.
Der Schlauch führt in einen Wasserbehälter, eine sogenannte pneumatische Wanne. Diese wird, mit Hilfe eines umgestülpten 100 mL Messkolbens, zur Messung des Gasvolumens verwendet.
Der Kolben ist über einer Hebebühne mit Magnetrührer angebracht. Es ist darauf zu achten, dass die Apparatur gerade aufgebaut und dicht ist. Die Schliffe müssen gefettet sein! Die 1,5%ige Wasserstoffperoxid-Lösung soll dabei aus der im Saal bereitstehenden 30%igen Lösung hergestellt werden.
Entfernen Sie das Thermometer und geben Sie einen Rührfisch in den Dreihalskolben. Befüllen Sie den Dreihalskolben mit 100 mL dest. H2O. Fügen Sie 3 mL der bereitgestellten Katalaselösung hinzu. Setzen Sie das Thermometer wieder ein und sichern Sie den Schliff. Versichern Sie sich, dass der Hahn des Tropftrichters geschlossen ist. Befüllen Sie den Tropftrichter mit 50 mL der vorbereiteten 1,5%igen Wasserstoffperoxidlösung. Achten Sie darauf, dass beim Befüllen keine Flüssigkeit über die Druckausgleichsleitung in den Dreihalskolben gelangt. Verschließen Sie den Tropftrichter mit einem Glasstopfen. Stellen Sie sicher, dass sich das Ende des Schlauches unter der Wasseroberfläche in der pneumatischen Wanne befindet. Stülpen Sie einen vollständig mit Wasser gefüllten Messzylinder über das Ende des Schlauchs, um das austretende Gas aufzufangen. Halten Sie eine Stoppuhr bereit.
Entscheiden Sie sich, bei welcher Temperatur Sie die Reaktion durchführen wollen (z.B. Raumtemperatur, 35°C, ~10°C, 4°C) und kühlen Sie ggf. auf die gewünschte Temperatur (Sprechen Sie sich mit Ihren Saalnachbarn ab, um sicherzustellen, dass Sie über Messdaten der Reaktion bei mindestens vier verschiedenen Temperaturen verfügen!).
Öffnen Sie, unter konstantem Rühren im Reaktionskolben, den Hahn des Tropftrichters. Starten Sie die Stoppuhr, sobald sich die Flüssigkeit komplett im Reaktionskolben befindet und notieren Sie 5-10 min lang alle 10 s den Wert des entstandenen Gasvolumens.
Die bereitgestellte Katalaselösung soll ohne weitere Verdünnung eingesetzt werden.
ACHTUNG: Die Apparatur muss nach der Versuchsdurchführung gründlich gereinigt werden!

Verändert ein Katalysator die Lage des chemischen Gleichgewichts einer Reaktion? [2P]
Tragen Sie die Messdaten in einem sinnvollen Schaubild auf. Beschreiben Sie den Verlauf der Kurve. [2P]
Bestimmen Sie die Reaktionsordnung der enzymatischen Zersetzung des Wasserstoffperoxids. Wie lautet das integrierte Geschwindigkeitsgesetz der Reaktion? [3P]
Einfluss der Temperatur auf die Reaktionsgeschwindigkeit
Die Geschwindigkeitskonstante k ist von der Temperatur T abhängig. Diese Abhängigkeit soll quantifiziert werden.
Kopieren Sie sich die
Ergebnisse der Messreihen bei den verschiedenen Temperaturen.
Stellen Sie die Messdaten von mindestens vier unterschiedlichen Temperaturen T in einem Schaubild dar. [1P]
Bestimmen Sie jeweils die Geschwindigkeitskonstante k bei den verschiedenen Temperaturen T. Welche Einheit hat die Geschwindigkeitskonstante? [3P]
Vergleichen Sie die k-Werte bei unterschiedlichen Temperaturen. Welchen Zusammenhang stellen Sie fest? [2P]
Wie kann die Zersetzung von Wasserstoffperoxid noch beschleunigt werden? Schätzen Sie ab, ob die anderen Katalysatoren mehr oder weniger effektiv als die Katalase arbeiten. [3P]
Stellen Sie verschiedenen k-Werte in einem Arrheniusdiagramm dar. Welche Größen werden an den Achsen jeweils aufgetragen?
Bestimmen Sie die Aktivierungsenergie EA der Reaktion. [5P]
Diese allgemeinen Konzepte sollte Sie hinter den Versuchen erkennen …
Der Ablauf einer chemischen Reaktion lässt sich mit Hilfe von mathematischen Gleichungen beschreiben. Die Reaktionsgeschwindigkeit spiegelt dabei die Konzentrationsabnahme der Edukte und die Konzentrationszunahme der Produkte pro Zeiteinheit wider. Die Reaktionsordnung beschreibt den Exponenten in der Gleichung.
Elektrochemie
In der Elektrochemie werden die Grundlagen für die Entwicklungen leistungsstarker Lithiumbatterien für Mobiltelephone und PCs, für Bleiakkumulatoren in Automobilen oder Brennstoffzellen zur Umwandlung von Methanol oder Wasserstoff in elektrische Energie in U-Booten, bzw. Raumstationen gelegt. Besonders das letztgenannte Arbeitsgebiet der Brennstoffzellen-Entwicklung ist dabei gegenwärtig als interdisziplinäres Feld der Physik, Chemie und den Materialwissenschaften aktuell. Auch in biologischen Systemen spielen elektrochemische Vorgänge eine bedeutende Rolle: man denke hierbei nur an die Atmungskette, die Cytochromoxidasen, die Erregungsleitung in Nervenzellen und viele weitere Vorgänge, die mit dem Verschieben von Elektronen und Ladungen auf zellulärer Ebene einhergehen. Elektrochemie ist dabei immer auch als Teilgebiet des wesentlich größeren Feldes der Chemie der Redoxreaktionen aufzufassen. Viele formale Dinge wie die Nernst'sche Gleichung oder das Aufstellen stöchiometrischer Redoxgleichungen finden sich deshalb hier in diesem Kapitel wieder.
Wissenswertes vorab
Aus dem Physik-Unterricht der Schule ist bekannt, dass elektrischer Strom modellhaft als Ladungstransport aufgefasst werden kann. Der Transport von Ladungen ist dabei an Ladungsträger wie Kationen, Anionen oder - im einfachsten Fall - Elektronen gebunden. Elektrischer Strom benötigt prinzipiell kein leitendes Medium, sondern kann auch im Vakuum erfolgen. Nachfolgende Versuche beschränken sich jedoch nur auf Vorgänge mit Ladungstransport in festen oder flüssigen leitenden Medien.
Die Beziehung zwischen Stromstärke I (SI-Einheit: Ampere, A), Spannung U (SI-Einheit: Volt, V) und elektrischem Widerstand R (SI-Einheit: Ohm, Ω) wird durch das Ohm'sche Gesetz beschrieben:
R = U/I
Bitte beachten: aus historischen Gründen fließt in der Sprache der Techniker der elektrische Strom vom Pluspol einer Stromquelle zum Minuspol („Technische Stromrichtung“, es werden also positiv geladene Ladungsträger bewegt), wohingegen der tatsächliche Stromfluss der Bewegung negativ geladener Elektronen zum Pluspol einer Stromquelle entspricht („Physikalische Stromrichtung“). Im Folgenden wird nur die physikalische Stromrichtung verwendet.
Wenn bei den Messungen eine konstante Spannung U verwendet wird, ändert sich laut dem Ohm'schen Gesetz die Stromstärke I nur durch einen veränderten Widerstand R. Der Widerstand wiederum ist invers proportional zur Leitfähigkeit der verwendeten Lösung. Deshalb verhält sich die gemessene Stromstärke proportional zur Leitfähigkeit der Lösung.
Die Beschreibung eines galvanischen Elements findet sich im Abschnitt „Bleiche, Desinfektion, oxidativer Stress: starke Oxidationsmittel“ des Skripts. Die folgende Abbildung zeigt als klassisches Beispiel einer galvanischen Zelle das Daniell-Element:
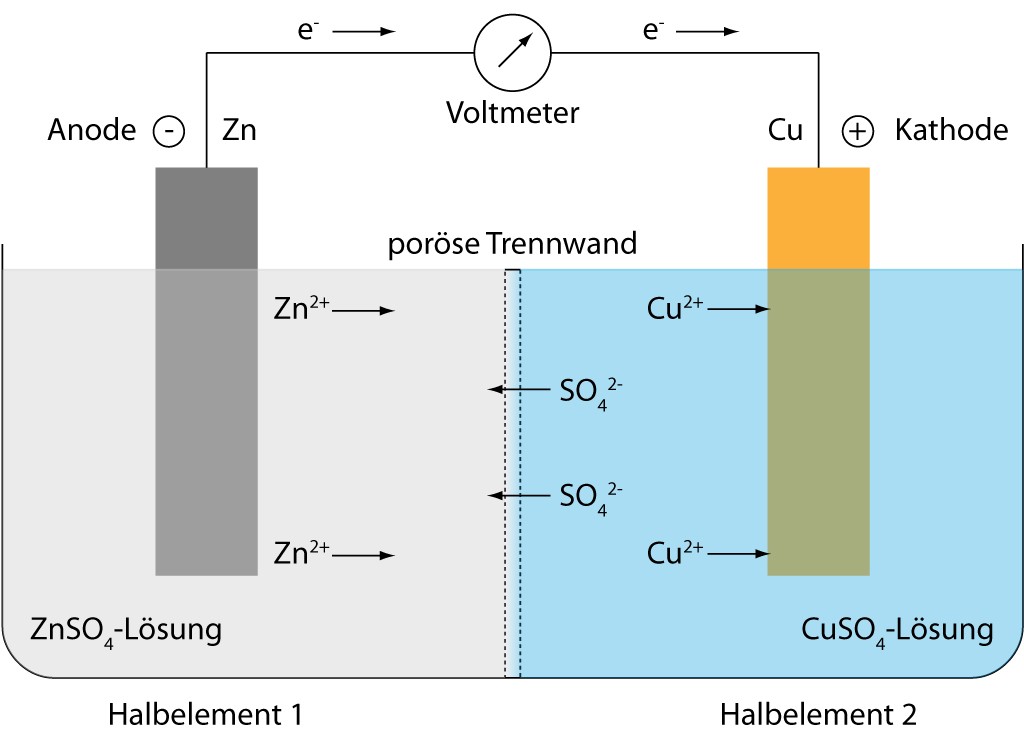
Es besteht aus einer Zinkhalbzelle die gegen eine Kupferhalbzelle geschaltet ist. Bei einem galvanischen Element ist die Anode der negative Pol an dem im Beispiel des Daniell-Elements elementares Zn zu Zn2+ oxidiert wird. Die Kathode ist der positive Pol an dem die Kupferionen zu elementarem Kupfer reduziert werden.
Es muss zwischen galvanischen Zellen und Elektrolysezellen unterschieden werden. Galvanische Zellen wandeln durch freiwillig ablaufende Reaktionen chemische Energie in elektrische Energie um. Bei Elektrolysezellen wird elektrische Energie zugeführt, um eine nicht freiwillig ablaufende chemische Reaktion in der Zelle zu erzwingen. Hierbei ist die Polung der Elektroden invers zur Polung in einer galvanischen Zelle. Die Anode ist der positive, die Kathode der negative Pol.
Aus dem Alltag sind galvanische Zellen als Batterien bekannt. Ist der Entladevorgang einer galvanischen Zelle durch Energiezufuhr reversibel spricht man von einem Akkumulator (Akku). Folglich können die Vorgänge beim Entladen eines Akkumulators als galvanische Zelle, beim Laden als Elektrolysezelle beschrieben werden.
Lernziele und einführende Literatur
Lernziele: Einschätzung der Leitfähigkeit wässriger Lösungen in Gegenwart bestimmter Ionen, Bestimmung der Polung einer Gleichstromquelle, elektrochemische Spannungsreihe und Konzentrationsgradienten als Ursache für Potentialgefälle, Bleiakkumulator, Maßanalytische Bestimmung am Beispiel der Konduktometrie und der Elektrogravimetrie
Einführende Literatur: Mortimer (Kapitel 20), Atkins (Kapitel 11 und 12)
Einführende Versuche
Die ersten Versuche sollen Ihnen ein Gefühl dafür vermitteln, welche Zusammenhänge zwischen Ladungstransport, Ladungsträgern und dem Stromfluss an sich sowie den dahinterliegenden und einhergehenden chemischen Vorgängen besteht. Nehmen Sie sich für die Versuche genügend Zeit! Da Messungen stets einen gewissen statistischen Fehler mit sich bringen, sollten Messungen mehrere Male durchgeführt werden, um diesen - zumindest ansatzweise - auszugleichen.
Leitfähigkeit von Ionen in wässriger Lösung
Lernziel: Sie sollen ein Gefühl dafür entwickeln, in welchem Umfang sich die Leitfähigkeit wässriger Lösungen in Abhängigkeit der Konzentration und Natur anwesender Ionen verändert.
Jede einzelne der hierzu benötigten wässrigen Salzlösungen soll von jeweils zwei Praktikanten angesetzt werden und kann dann im Tausch von allen Kursteilnehmern im Saal verwendet werden. Die Lösungen sollten bereits am Vortag bzw. in der Vorwoche hergestellt werden, um die Einstellung der Lösungen auf Zimmertemperatur sicherzustellen. Mittels der Messkolben (250 mL) werden jeweils 0,1 m Lösungen der Salze aus der untenstehenden Tabelle hergestellt (beachten Sie bei Ihren Berechnungen, dass einige der Salze in kristalliner Form als definierte Hydrate vorliegen).
Ferner sollen ausgehend von konzentrierter Salzsäure bzw. festem Natriumhydroxid 0,1 m Lösungen von Salzsäure und Natronlauge hergestellt werden.
Der Versuchsaufbau besteht aus einem Konduktometer mit einer Leitfähigkeitselektrode.
Alle Salzlösungen sollen auf ihre jeweiligen Leitfähigkeiten hin überprüft werden. Zweckmäßigerweise geht man hierzu folgendermaßen vor: das Konduktometer wird eingeschaltet und vor der ersten Verwendung mit den bereitgestellten Kalibrierlösungen kalibriert. Die Leitfähigkeitselektrode wird in die zu messende Flüssigkeit eingetaucht und der angezeigte Wert abgelesen. Vor jeder Messung müssen die Elektroden gründlich mit deionisiertem Wasser abgespült werden. Als letztes soll die Leitfähigkeit der Natronlauge und der Salzsäure bestimmt werden.
| Kat+ | Kat2+ | A- | A2- | |
|---|---|---|---|---|
| LiCl | MgCl2 | NaF | Na2SO4 | NaOH |
| NaCl | BaCl2 | NaI | Na2C2O4 | HCl |
| KCl |
Starke und schwache Elektrolyte Katx+Ay- mit unterschiedlicher Ionenstärke.

Tragen Sie die ermittelten Stromstärken tabellarisch auf und diskutieren Sie die Änderung der Leitfähigkeit in der Reihe der einwertigen, zweiwertigen und dreiwertigen Kationen sowie der einwertigen und zweiwertigen Anionen. [3P]
Erklären Sie die überraschend gute Leitfähigkeit der Natronlauge und der Salzsäure im Vergleich zur ungesättigten Natriumchlorid-Lösung. [2P]
Welche Veränderung der Leitfähigkeit würden Sie bei einer gesättigten Salzlösungen erwarten? Begründen Sie Ihre Annahmen. [2P]
Bestimmung der Polung einer Gleichstromquelle
Lernziel: Stromfluss durch wässrige Lösungen ist in vielen Fällen mit einer Veränderung des pH-Werts verbunden. In einer hierfür geeigneten Versuchsanordnung kann dieser Effekt zur Bestimmung der Polung einer Gleichstromquelle (z.B. einer Batterie) benutzt werden.
Die beiden Pole einer 4,5-Volt-Batterie werden jeweils mit einem Draht verlängert (welche zuvor gründlich mit Wasser und Seife gereinigt wurden). Anschließend wird eine etwa 0,1 m wässrige Lösung von Natriumchlorid hergestellt und mit einigen Tropfen Phenolphthalein-Lösung versetzt. Mit dieser wird ein Streifen Filterpapier (etwa 5 × 1 cm) getränkt, welcher danach auf einem Uhrglas ausgebreitet wird.
Vorsicht: der Filterpapierstreifen soll für dieses Experiment zwar feucht sein, auf dem Uhrglas soll jedoch keine Pfütze stehen!
Die Enden der beiden Drähte werden in einem Abstand von etwa 2 cm nebeneinander auf den Filterpapiertstreifen gepresst.
Notieren und erklären Sie alle Beobachtungen und formulieren Sie die ablaufenden Reaktionen. Wie kann daraus eindeutig die Polung der Batterie abgeleitet werden? [2P]
Wiederholen Sie den Versuch mit einem neuen Filterpapierstreifen und stellen Sie dieses Mal einen Abstand von 4 cm zwischen den beiden Drähten auf dem Filterpapier ein.
Wie ist der zeitlich andere Verlauf der Beobachtungen zu erklären? [1P]
Galvanische Zellen
Lernziel: Wie wird in einer Batterie überhaupt elektrischer Strom erzeugt? Sie sollen erkennen, dass durch den Kontakt verschiedener Metalle und Metallionen Potentialgefälle erzeugt werden. Das Ausmaß des Potentialgefälles hängt dabei sowohl von der Konzentration der beteiligten Reaktionspartner als auch von der unterschiedlichen Stellung der Reaktanden in der elektrochemischen Spannungsreihe ab.
Bleinitrat ist als fruchtschädigend eingestuft. Schwangere sollten daher in keinem Fall Umgang mit Bleinitrat haben. (Kann das Kind im Mutterleib schädigen. Kann möglicherweise die Fortpflanzungsfähigkeit beeinträchtigen. H-Sätze: H360Df/302+332/318/372/410. Bitte beachten Sie auch das Sicherheitsdatenblatt).
Zunächst werden 0,1 m wässrige Lösungen von Bleinitrat, Zinknitrat und Kupfernitrat vorbereitet und in Bechergläser (100 mL) gefüllt. Anschließend werden zwei Streifen Kupferblech, Zinkblech und Bleiblech gründlich mit Wasser und Seife gereinigt (Vorsicht vor scharfen Kanten!), mit einigen Tropfen Salpetersäure (6 M) und Zellstoff von anhaftenden Verunreinigungen befreit, erneut gründlich mit Wasser gespült (blankes, glänzendes Metall muss sichtbar sein!) und in die zugehörige Salzlösung gestellt. Diese Anordnung wird jeweils als „Halbelement“ bezeichnet.
Um zwischen beiden Halbelementen eine ionenleitende Verbindung zu ermöglichen, verwenden Sie Salzbrücken. Diese werden jetzt aus den bereitgestellten U-förmig gebogenen Glasrohren gebaut, indem man diese vollständig mit wässriger 0,5 m Ammoniumnitrat-Lösung füllt und an beiden Enden mit einem Wattepfropf luftblasenfrei verschließt. Man verwendet zunächst die Glasrohre mit einem mittleren Innendurchmesser (etwa 7 mm).
Zur Messung der sich zwischen zwei Halbelementen ausbildenden Spannung verbindet man die Metallblechstreifen zweier nebeneinanderstehender Halbelemente mit einem Voltmeter (Krokodilklemmen verwenden), verbindet die beiden Halbelemente durch Eintauchen der Salzbrücke und liest sofort die Spannung ab. Nach der Messung wird die Salzbrücke wieder aus den beiden Halbelementen herausgezogen. Um eine Messung mehrfach zu wiederholen, merkt man sich, welches der Enden mit welcher Salzlösung in Kontakt stand, da diese beim neuerlichen Eintauchen keinesfalls vertauscht werden dürfen.
Folgende Teilexperimente sollen in der angegebenen Reihenfolge durchgeführt werden:
• Spannungsmessung unter Verwendung unterschiedlicher Elektrodenpaare (Pb/Zn, Pb/Cu und Cu/Zn; s. a. Frage 3.6 und 3.7)
• Spannungsmessung bei unterschiedlicher Eintauchtiefe (unterschiedliche Metallaktivität) der Elektroden (s. a. Frage 3.9)
• Spannungsmessung mit Cu/Cu Element in unterschiedlich konzentrierten Kupferlösungen (zuerst 0,1 m / 0,1 m, dann 0,1 m / > 0,1 m) (s. a. Frage 3.10)
Was folgt aus den erhaltenen Werten der Spannungsmessung der Paare Pb/Zn und Pb/Cu über die Einordnung der drei Metalle relativ zueinander in der elektrochemischen Spannungsreihe? Welche Spannung kann man deshalb für das Paar Cu/Zn erwarten? Wird dieser Wert auch experimentell erhalten? [2P]
Vergleichen Sie Ihre Messergebnisse mit den erwarteten Werten, die sich aus tabellierten Normalpotentialen ergeben hätten. Was könnten die Ursachen für Abweichungen sein? [2P]
Welches Verhalten der Abweichung zwischen dem theoretischen und dem experimentellen Wert erwarten Sie für Salzbrücken mit kleinerem und größerem Innendurchmesser? Wie könnte man folglich die Abweichung noch weiter minimieren? Was wäre anstelle einer Salzbrücke idealerweise zu verwenden? [3P]
Die Berechnung der Potentiale nach der Nernst'schen Gleichung erfolgt unter der Annahme konstanter Metallaktivität, welche willkürlich auf den Wert 1 gesetzt wird. Stimmt diese Näherung? Verändern Sie durch mehr oder weniger tiefes Eintauchen der Blechstreifen in die Metallsalzlösungen die wirksame Metalloberfläche und überprüfen Sie, ob sich die Spannung dabei ändert. [2P]
Ersetzen Sie nun im Cu/Zn-Paar den Zinkblechstreifen durch einen Kupferblechstreifen, wodurch der Cu-Elektrode jetzt eine weitere Cu-Elektroden gegenübersteht. Welche Spannung wird jetzt zwischen den beiden Halbelementen gemessen? Was für eine Anordnung liegt jetzt vor? Wie könnte man die Spannung dieser Cu/Cu-Zelle noch weiter erhöhen? Überprüfen Sie experimentell Ihre Vermutung, indem Sie die 0,1 m Kupfernitrat-Lösung durch eine andere Lösung mit geeigneter Kupferkonzentration austauschen. [3P]
Bleiakkumulator
Lernziel: Galvanische Elemente, in denen sich durch das Anlegen einer äußeren Spannung die bei der Stromentnahme ablaufenden Reaktionen umkehren lassen, bezeichnet man als „Akkumulatoren“. Am Beispiel des in der Automobilindustrie wichtigen Bleiakkumulators sollen Sie die Funktionsweise – insbesondere das Entladen und Aufladen – eines Bleiakkumulators kennenlernen.
Als Elektroden dienen im Bleiakkumulator Blei und eine Bleidioxidelektrode, als Elektrolyt wird Schwefelsäure (20%) verwendet. Bei der Entnahme von Strom wird die Bleielektrode zu Bleisulfat oxidiert und fungiert als negativer Pol des Akkumulators. Am positiven Pol wird Bleidioxid zu Bleisulfat reduziert. Formulieren Sie die Teilgleichungen an beiden Polen des Bleiakkumulators bei Stromentnahme sowie die Gleichung für die Gesamtreaktion. [3P]
Was wird letztlich also beim Entladen eines Bleiakkumulators verbraucht? In Autowerkstätten wird dieser Umstand zur Bestimmung des Ladungszustands eines solchen Akkumulators ausgenutzt. Was genau wird dabei gemacht, worauf beruht diese Bestimmungsmethode? [3P]
Zwei Bleiblechstreifen werden – wie oben für die Kupferblechstreifen beschrieben – gereinigt und für eine Stunde in 1,0 m Schwefelsäure gehängt. Die sich dabei bildende dünne Bleisulfatschicht führt dazu, dass die Elektroden nach diesem Zeitraum einheitlich matt-grau erscheinen. Mittels Krokodilklemmen werden die beiden Streifen anschließend für fünf Minuten mit den Polen einer Gleichstromquelle von 1 Volt Spannung verbunden und in die 20%ige Schwefelsäure auf gleiche Höhe eingetaucht.
Wie ändert sich während dieses Ladevorgangs das Aussehen der beiden Bleiblechstreifen? [2P]
Formulieren Sie die Gleichungen für die an beiden Polen des Akkumulators ablaufenden Reaktionen beim Ladevorgang. [3P]
Unterbrechen Sie die Stromzufuhr und bestimmen Sie die Spannung zwischen den Polen des geladenen Akkumulators mit Hilfe eines Multimeters.
Bestimmen Sie die Spannung zwischen den Polen Ihres jetzt frisch geladenen Akkumulators mit Hilfe eines Multimeters und geben diese an. [1P]
Wiederholen Sie den Ladevorgang, verwenden Sie dieses Mal jedoch eine Spannung von 5 Volt.
In welchem Zeitraum können jetzt Veränderungen an den Bleiblechstreifen beobachtet werden? [1P]
Was kann jetzt außerdem an den Elektroden beobachtet werden? Um welches Reaktionsprodukt handelt es sich dabei? Formulieren Sie alle notwendigen Reaktionsgleichungen! [3P]
Warum wird in der Praxis einer Autowerkstatt also das Laden eines Bleiakkumulators nicht mit beliebig hoher Spannung (sagen wir 1000000 Volt) durchgeführt, obwohl es doch dann so viel schneller ginge? [1P]
Vollanalyse Konduktometrie
Die in den Eingangsversuchen beobachtete Änderung der elektrischen Leitfähigkeit wässriger Lösungen in Abhängigkeit von den anwesenden Ionen kann zur maßanalytischen Bestimmung von Säuren und Laugen und sogar von Mischungen unterschiedlich starker Säuren verwendet werden. Die außergewöhnlich gute Leitfähigkeit von Hydronium-Ionen und Hydroxid-Ionen im Vergleich zu den nach Neutralisation anwesenden Ionen stellt die Basis der Konduktometrie dar. Im Folgenden soll aus einer Mischung einer starken Mineralsäure (Salzsäure) und einer schwachen organischen Säure (Essigsäure) mittels dieser Methode der Gehalt der beiden Einzelkomponenten bestimmt werden. Der Gesamtgehalt an Säure im Gemisch liegt im Intervall von 150-250 mg, wobei sich die Anteile der beiden Säuren deutlich voneinander unterscheiden können.
Da zur Bestimmung der Gehalte nur die Änderung der relativen, nicht aber der absoluten Leitfähigkeiten von Bedeutung ist, müssen die Konduktometer nicht eingangs kalibriert werden. 50 mL der ausgegebene Analysen-Lösung wird in ein hohes Becherglas gegeben und die Elektroden des Konduktometers werden in das Glas eingesenkt. Unter Rühren mit dem Magnetrührer wird das Volumen der Analysenlösung solange mit deionisiertem Wasser vergrößert, bis die Elektroden sicher in die Lösung eintauchen. Anschließend lässt man unter fortgesetztem Rühren aus einer Bürette Kalilauge (Natronlauge ist weniger gut geeignet) bekannter Konzentration in Anteilen von jeweils 1,0 mL zufließen und notiert nach einer Wartezeit von jeweils einer Minute die Leitfähigkeit der Lösung. Nach Möglichkeit sollte das Volumen einer 25-mL-Bürette vollständig ausgenutzt werden. Es bringt keinen Nutzen, um den oder die erahnten Äquivalenzpunkte herum kleinere Titervolumina zur Maßlösung zu geben, da in diesem Bereich die Leitfähigkeit erfahrungsgemäß größeren Schwankungen unterliegt.
Die erhaltenen Werte werden zunächst notiert. Die Erstellung der Leitfähigkeitskurve und die Auswertung können über das Programm „QtiPlot“ erfolgen. Zur Bestimmung der Äquivalenzpunkte hat es sich als vorteilhaft erwiesen, nur die „eindeutigen“ Punkte zu berücksichtigen, d.h. es sollen nur durch die Punkte im Bereich des kontinuierlichen Abfalls der Leitfähigkeit, des langsamen sowie des raschen Wiederanstiegs der Leitfähigkeit Ausgleichsgeraden gelegt werden. Aus den Koordinaten des ersten Schnittpunktes kann das zur Neutralisation der Salzsäure notwendige Volumen an Kalilauge sicher bestimmt werden. Aus den Koordinaten des zweiten Schnittpunkts lässt sich der Gesamtverbrauch an Natronlauge zur Neutralisation ermitteln, die Differenz zum Äquivalenzpunkt der Salzsäure ergibt den Verbrauch zur Neutralisation der Essigsäure.
Stellen Sie Ihre gemessene Leitfähigkeitskurve graphisch dar. [2P]
Geben Sie den Gehalt der beiden Säuren in Ihrer Maßlösung an. [2P]
Machen Sie sich das Zustandekommen der Leitfähigkeitskurven nochmals klar– wieso verläuft die Leitfähigkeit in der angegebenen Weise, obwohl sich während der gesamten Titration die Zahl der in Lösung vorhandenen Ionen nicht ändert? [2P]
Vollanalyse Elektrogravimetrie
Die Elektrolyse von Metallsalzlösungen und -schmelzen stellt ein wichtiges Reinigungsverfahren für Metalle und in einigen Fällen die Hauptdarstellungsmethode elementarer Metalle dar. Mittels elektrolytischer Verfahren kann auch der Gehalt einer Lösung an Metall-Ionen gravimetrisch bestimmt werden. Besonders gut verläuft diese Bestimmung bei edleren Metallen wie Kupfer, deren Ionen sich leicht an einer negativ geladen Elektrode abscheiden lassen.
Die Versuchsanordnung besteht aus einer Elektrolysestation und zwei Platinelektroden in Form eines Drahtnetzes und eines spiralförmig gebogenen Drahtes. Zunächst sollten Sie sich mit der Funktionsweise der Elektrolysestation vertraut machen und die Polung der Gleichstromquelle überprüfen (das Rüstzeug dazu haben Sie in einem der Vorversuche erhalten). Machen Sie sich klar, an welchem Pol das Drahtnetz und an welchem Pol die Platinspirale angeschlossen werden muss.
Das Platinnetz und der Platindraht werden zunächst mit heißer konzentrierter Salpetersäure, welche keine Spuren von Chlorid enthalten darf, gereinigt. Die Säure wird dazu am besten zentral für alle anwesenden Praktikanten in einem größeren Becherglas auf etwa 100 °C erhitzt und die Platinelektroden werden für etwa eine Minute eingetaucht. Anschließend spült man der Reihe nach sehr gründlich mit deionisiertem Wasser und absolutem Alkohol (Achtung! Nicht über der konzentrierte Salpetersäure spülen und danach auf keinen Fall wieder in die Säure eintauchen!) und trocknet bei 110 °C im Trockenschrank für etwa 30 Minuten. Nach dem Abkühlen auf Raumtemperatur bestimmt man an der Analysenwaage die Masse der Drahtnetzelektrode. Anschließend wird der spiralförmige Platindraht als Anode, die Elektrode in Form des Drahtnetzes als Kathode in das Elektrolysegerät eingebaut. Der spiralförmige Draht wird dabei vollständig vom Netz umgeben, darf dieses jedoch keinesfalls berühren (auch beim späteren Rühren nicht!).
Ein Aliquot (50 mL) der auf die übliche Weise vorbereiteten Analysenlösung wird in ein hohes Becherglas (150 mL) gegeben, mit 10 mL Schwefelsäure (20%) versetzt und auf ein Gesamtvolumen von 70–80 mL verdünnt. Die Elektroden sollen dabei nur etwa zur Hälfte in die Probenlösung eintauchen. Man erwärmt den Inhalt des Becherglases unter Rühren mit einem Rührfisch auf 70°C und elektrolysiert bei einer Spannung von 2,5–3,0 Volt. Nach etwa 20 Minuten erhöht man den Flüssigkeitsstand etwas durch Zugabe von deionisiertem Wasser und überprüft, ob sich an der jetzt neu in die Lösung eintauchenden Fläche im Laufe einiger Minuten noch etwas Kupfer abscheidet. Der Vorgang wird ggf. solange wiederholt, bis sich auch 15 Minuten nach Zugabe von deionisiertem Wasser unter fortgesetzter Elektrolyse keine Kupferabscheidung mehr beobachten lässt.
Nach beendeter Elektrolyse werden die Elektroden aus der Probenlösung gezogen und (noch unter Spannung) mit deionisiertem Wasser abgespült. Anschließend wäscht man diese noch mit reinem Alkohol, trocknet im Trockenschrank bis zur Gewichtskonstanz und bestimmt erneut die Masse.
Geben Sie den Gehalt an Kupfer in Ihrer Maßlösung an. [1P]
Bis zu welcher Restkonzentration kann man die Kupfer-Abscheidung aus saurer Lösung (pH = 1) theoretisch beobachten, bevor an der Kathode Wasserstoff entwickelt wird? Geben Sie Ihren Rechenweg an (E0(Cu/Cu2+) = 0,345 V) [3P]