 Erst 'mal tief Luft holen? – Unser gefährliches Leben als Aerobier
Erst 'mal tief Luft holen? – Unser gefährliches Leben als Aerobier Freitag, 22. Februar 2008, 17:30–19:00, Vortragssaal der Wacker AG, Burghausen anlässlich der Chemie-Olympiade 2008.
Die Versuche wurden von Chemotechniker Arthur Gruber entwickelt und vorbereitet.
Derzeit sind ca. 300 Enzyme strukturell aufgeklärt. Eine Strukturanalyse in atomarer Auflösung bietet den notwendigen Rahmen zur Analyse des Reaktionsverlaufs im aktiven Zentrum des Enzyms. Ebenso wie in der technischen Katalyse finden sich auch in den aktiven Zentren von Enzymen Metallzentren als Ort der katalytischen Umsetzung des jeweiligen Substrats. Die besonders reiche Chemie von Metallen – vor allem von Übergangsmetallen – ist die offensichtliche Ursache dafür, dass Metalle in den aktiven Zentren von Enzymen von viel größerer Bedeutung sind als anderswo in der Biochemie: etwa die Hälfte der strukturell aufgeklärten Enzyme sind metallhaltige Proteine!
Sauerstoff ist der reaktive Luftbestandteil:
Versuch 8-1: Fe zeigt 1/5 O2 in Luft an • mit Essigsäure gewaschene Eisenwolle entzieht der Luft einen Bestandteil; die Reaktion kommt zum Erliegen, wenn ca. 1/5 des eingesetzten Luftvolumens verbraucht ist. Luft ist also ein Gasgemisch, das zu ca. 1/5 aus einer reaktiven Komponente besteht, genauer zu 21 Volumen-% aus Sauerstoff, und zu ca. 4/5 aus einem Rest, der unter diesen Bedingungen nicht reagiert, nämlich 78 Vol.-% Stickstoff und 1 Vol.-% Argon.
Sauerstoff oxidiert bei diesem Versuch elementares Eisen zur Oxidationsstufe +III:
4 Fe + 3 O2 → FeIII2O3
Die dreiwertige Stufe des Eisen wird auch ausgehend von einer Eisen(II)-Verbindung in Wasser durch Luftsauerstoff bei Raumtemperatur erreicht:
4 FeII(OH)2 + O2 + H2O → 4 FeIII(OH)3
Reaktionen dieses Typs führten nach der Anreicherung von Sauerstoff in der Atmosphäre durch die photosynthetische Aktivität von Braunalgen vor ca. 2–3 Milliarden Jahren zur Oxidation von Eisenvorkommen. Natürliche Eisen(III)-oxid-Vorkommen sind bisweilen sehr auffällig, so dass das schwere Mineral Fe2O3, „Hämatit“, bereits in der frühen Antike als wertvoller Rohstoff erkannt wurde – die „Eisenzeit“ begann.

Ein ca. 1 m großer Eisenoxidbrocken am Monte Calamita im Südosten Elbas (ital. calamita, Magnet). Das Vorkommen wurde beginnend mit der Etruskerzeit bis zum 2. Weltkrieg ausgebeutet.
Eine wichtige Eigenschaft von Sauerstoffmolekülen ist deren Paramagnetismus, dessen Betrag zwei ungepaarte Elektronen pro O2-Molekül anzeigt. Diese Eigenschaft lässt sich an flüssigem Sauerstoff demonstrieren, einer blauen Flüssigkeit (links, rechts zum Vergleich flüssiger Stickstoff).

Versuch 8-13: Kondensation von flüssigem Sauerstoff an stickstoffgekühltem Kupferfinger
Versuch 8-10: Flüssiger Sauerstoff ist blau, flüssiger Stickstoff farblos
Versuch 8-11/9-1: Paramagnetismus von Sauerstoff • ein mit flüssigem Sauerstoff gefülltes Reagenzglas wird deutlich in das Feld eines Elektromagneten hineingezogen) und Diamagnetismus von Stickstoff (ein mit flüssigem Stickstoff gefülltes Reagenzglas scheint unter diesen Bedingungen nicht auf das Magnetfeld zu reagieren.
Der Paramagnetismus von Sauerstoff ist nicht unerwartet, denn der Versuch, die Bindungen im O2-Molekül aus den Orbitalen zweier O-Atome zu bilden, führt zu einer problematischen Situation:
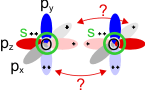
Obwohl geometrisch gleichwertig, sind die px- und py-Orbitale jetzt unterschiedlich besetzt – eines mit einem einzelnen Elektron, das andere mit einem Elektronenpaar.
Eine Molekülorbitalrechung berücksichtigt dei Äquivalenz der px- und py-Orbitale. Mit der Valenzelektronenzahl von 2 × 6 = 12 ist der 2π-Zustand mit zwei Elektronen besetzt und zwar wegen der Hundschen Regel mit jeweils 1 Elektron – O2 ist paramagnetisch. Die Beschreibung des Spinzustands ist als „Multiplizität“ gebräuchlich. Hierzu werden die Elektronenspins von ½ zu einem Gesamtspin S addiert, anschließend wird die Multiplizität als 2S + 1 gebildet (für das Sauerstoffmolekül mit S = 2 × ½ = 1: 2 × 1 + 1 = 3). Teilchen mit einer Multiplizität von 1, 2, 3 sind im Singulett-, Dublett-, Triplett-Zustand. Die stabile Form von O2 ist also der Triplett-Sauerstoff.
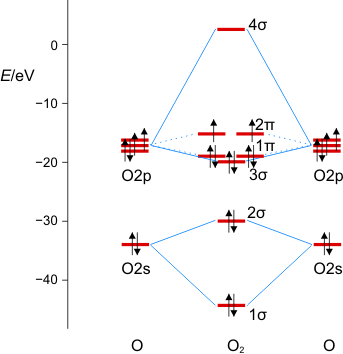
MO-Diagramm für O2.
Technische Anmerkung: O2-Energien und Atomabstand mit ump2/aug-cc-pvqz, 1.219 Å Atomabstand.
Sauerstoff, ein „Diradikal“, gehört zu den wenigen Molekülen, die stabil sind, ohne dass sich im Grundzustand für die Summe der Spinquantenzahlen 0 ergibt, ohne also dass alle Elektronen spingepaart vorliegen. Zahlreiche chemische Reaktionen laufen nur ungehemmt ab (meist solche, bei denen keine Übergangsmetalle anwesend sind), wenn die Summe der ungepaarten Spins gleich bleibt. Entsteht bei einer solchen Reaktion Sauerstoff, so wird dieser nicht im Triplett-Grundzustand frei, sondern als unstabiler und äußerst reaktiver Singulett-Sauerstoff, der eine elektronisch angeregte Form darstellt.
Versuch 9.2: Singulettsauerstoff • Bei der Reaktion
H2O2 + Cl2 + 2 OH− → 1O2 + 2 Cl− + 2 H2O
wird Singulett-Sauerstoff gebildet. Bei der Rückkehr in den 3O2-Grundzustand wird rotes Licht der Wellenlängen 633 und 760 nm emittiert (190 = 2 × 95 und 158 kJ mol−1 entsprechend einer Emission zweier Singulett-Sauerstoff-Formen), hierzu Holleman-Wiberg: „… ist die Umsetzung von Hypochlorit mit Wasserstoffperoxid von einer Emission begleitet, die man mit dunkeladaptiertem Auge als roten Schimmer wahrnehmen kann.“
Hr ist sowohl im unbeladenen als auch im O2-beladenen Zustand kristallographisch charakterisiert. Hr ist ein kleines Protein mit 113 Aminosäuren, die in charakteristischer Weise einen Stapel aus vier α-Helices bilden. Gezeigt ist dies am Beispiel des Hämerythrins von Themiste dyscritum, eines im Meer lebenden Wurms, das im beladenen und unbeladenen Zustand analysiert wurde. Abgebildet sind zwei Ansichten des Säulenstapels der unbeladenen Form:


Das aktive Zentrum weist eine freie Koordinationsstelle auf; die Ligandausstattung besteht aus fünf His-Seitenketten, einem verbrückenden Hydroxo-Ligand, 1 Asp und 1 Glu:

An diese Position bindet im beladenen Zustand das O2-Molekül

Quantenchemische Rechnungen zeigen, dass die Sauerstoff-Anlagerung ein Redox-Prozess ist, der durch eine Protonenverschiebung begleitet wird: Durch den Übertrag von zwei Elektronen entstehen zwei Eisen(III)-Zentren und ein Peroxid-Ion. Das Anion nimmt das Proton des verbrückenden Hydroxo-Liganden auf und betätigt eine Wasserstoffbrückenbindung zum gebildeten Oxo-Ligand. Bei der Freisetzung verlaufen die umgekehrten Vorgänge, Eisen(III) muss Peroxid zu Sauerstoff oxidieren können. Das Standardpotential in saurer Lösung für die Peroxid-Oxidation beträgt 0.7 V, bei pH 7 ergibt sich 0.3 V. Ohne Liganden beträgt das Eisen(II,III)-Potential bei pH 7 ca. 0 V, eine Sauerstoff-Freisetzung wäre unter diesen Bedingungen nicht möglich. Hr ist ein weiteres Beispiel, wie eine größere Zahl von His-Liganden das elektrochemische Potential so weit erhöht, dass die benötigte Oxidationskraft der oxidierten Form zur Verfügung steht.
Der Weg des Sauerstoffs von der Lunge bis in die Mitochondrien beginnt mit der Bindung an Hämoglobin. In den Muskelzellen erfolgt die Übergabe an Myoglobin, das eine größere Bindungskonstante für O2 als Hb hat. Von diesem wird das Sauerstoffmolekül schließlich an Cytochrom-c-Oxidase (CcO) übertragen, bei dem die Sauerstoffaffinität den größten Wert erreicht. Im aktiven Zentrum von CcO, dem letzten Enzym der Atmungskette, wird Sauerstoff zu Wasser reduziert. Die Sauerstofftransportenzyme Hb und Mb kommen bei allen Wirbeltieren und vielen Wirbellosen vor. Das Mengenverhältnis von Mb zu Hb ist zwischen den Organismen sehr verschieden. Besonders große Mb-Mengen werden von im Wasser lebenden lungenatmenden Tieren genutzt, um viel Sauerstoff für lange Tauchgänge speichern zu können. So können Wale und Robben aufgrund ihres hohen Mb-Spiegels ca. ½ h unter Wasser bleiben. Das in der Biochemie allgegenwärtige Pottwal-Mb (engl. sperm whale myoglobin) erklärt sich aus der hohen Verfügbarkeit dieses Proteins.
Beide Transportenzyme sind nur dann in der Lage Sauerstoff zu binden, wenn das Eisenzentrum in der Oxidationsstufe +II vorliegt (deoxyMb und deoxyHb). Oxidation zur dreiwertigen Stufe unter Bildung von metMb oder metHb führt zur Desaktivierung.
Eine neu entdeckte Funktion von Enzymen der Mb- und Hb-Familie scheint die Beseitigung von NO zu sein. Mbs and Hbs können NO zu Nitrat oxidieren. Ausgehend von oxyMb oder oxyHb, deren FeII-O2-Fragment sich hierbei wie eine FeIII-O2•−-Funktion verhält (siehe auch weiter unten), lässt sich die Umsetzung gemäß
NO + O2•− → NO3−
formulieren, wobei metMb oder metHb zurückbleibt und anschließend durch eine 1-e-Reduktion wieder in den zweiwertigen Ausgangszustand überführt werden muss [heme4].
Häm-Proteine traten in der Evolution weit vor dem Aufkommen von Sauerstoff in der Atmosphäre auf. Die NO-bindenden Eigenschaften haben zu der Hypothese geführt, dass das „Ur-Hämoglobin“, dessen Entstehung vor ca. 3.5 Milliarden Jahren vermutet wird, dem NO-Stoffwechsel diente und dass der O2-Transport eine erst später evolvierte Eigenschaft ist.
Mb ist ein 153 Aminosäuren langes Protein, das durch acht α-Helices dominiert ist. Eine hydrophobe Tasche des Apoproteins enthält einen Eisen(II)-Protoporphyrin-IX-Komplex (Häm b) ohne kovalente Anbindung des Porphyrins nur über einen His-Fe-Kontakt (siehe Abbildung bei oxyMb). Das unmittelbar an Eisen gebundene His ist das „proximale“ His (His93, oft als F8 adressiert [8. Aminosäure auf α-Helix F]). Auf der anderen Häm-Seite liegt die O2-Bindungsstelle. Hier wird in der Regel ein weiteres His gefunden, das „distale“ His (His64 oder E7), das in passender Entfernung zum Häm lokalisiert ist, um als N-H-Donor eine Wasserstoffbrückenbindung zu H-Brücken-Akzeptoren aufbauen zu können.
Das Bild zeigt deoxy-Mb bei hoher Auflösung (1 Å). Das distale His wird fehlgeordnet gefunden, das auf der distalen Seite eingezeichnete Wassermolekül ist in unterbesetzter Lage. Aufgrund der für Wasserstoffbrückenbindungen typischen N···O-Abstände zwischen der Wasserlage und dem distalen His (2.76 Å) sowie der Wasserlage und einem der vier Porphyrin-Stickstoffatome (2.74 Å) lässt sich die Fehlordnung des distalen His verstehen: in einem Teil der den Kristall bildenden Mb-Moleküle fehlt das einzelne Wassermolekül auf der distalen Seite. Dies ist verständlich, da es außer den beiden N···O-Kontakten keine weiteren Bindungsmöglichkeiten für den Wasser-Dipol gibt – der distale Hohlraum ist außer von His nur von hydrophoben Seitenketten ausgekleidet (siehe unten). Das distale His nimmt die linke Position aber nur bei Anwesenheit von Wasser ein. Fehlt das Wassermolekül, bewegt sich das distale His nach rechts in Richtung auf den Proteinrand, wo es einen nicht gezeigten Kontakt zu einem Wassermolekül außerhalb der O2-Bindungstasche aufbauen kann.

Das Eisen(II)-Zentrum in deoxyMb liegt im S=5/2-Grundzustand vor, es ist ein high-spin-Zentrum.
Auch die Struktur von oxyMb liegt in hoher Auflösung vor. Der Blick auf das Holoenzym zeigt die Lage des Häms in einer von α-Helices gebildeten Tasche. Die α-Helices sind in der üblichen Weise bezeichnet:

Im aktiven Zentrum ist ein Sauerstoffmolekül gebunden, dessen weitere Umgebung mit Ausnahme des distalen His nur aus hydrophoben Seitenketten besteht:

Die Wechselwirkung zwischen dem distalen His und O2 wird deutlicher, wenn der Betrachter von der linken Seite in das aktive Zentrum hineinschaut:

Das distale His ist Donor einer Wasserstoffbrückenbindung zum terminalen O-Atom des O2-Liganden. Die N–H···O-Bindung ist nicht die Ursache der gewinkelten Fe-O-O-Anordnung, sie nutzt diese jedoch zum Aufbau der zusätzlichen Wechselwirkung und erhöht so die Bindungskonstante für O2.
oxyMb liegt im Singulett-Grundzustand vor, was Pauling zu der Schlussfolgerung veranlasste, oxyMn sei ein low-spin-Eisen(II)-Komplex.
Die Stabilisierung des O2-Komplexes durch die H-Brückenbindung zum distalen His scheint der wesentliche Grund für die relativ geringe CO-Affinität von Mb und Hb zu sein. Während isoliertes Häm CO ca. 105 mal wirksamer bindet als O2, so ist dieses Verhältnis bei Mb und Hb um den Faktor 1000 geringer. Eine dann 100-fache Affinität zu CO weist Kohlenmonoxid immer noch als giftiges Gas aus, wenn es in unphysiologisch hoher Konzentration eingeatmet wird. Die zum Beispiel beim Häm-Abbau entstehende geringe physiologisch gebildete CO-Menge jedoch verliert bei diesem Verhältnis der Bindungskonstanten die Fähigkeit, Mb oder Hb zu blockieren.
Es liegen zahlreiche Strukturanalysen zur Bindung von CO an Mb vor. Die Leu29Trp-Mutante von Pottwal-Mb, bei welcher der hydrophobe Raum auf der distalen Seite durch die Mutation verkleinert ist, wurde detailliert untersucht. Das CO-Addukt zeigt ein nur geringfügig abgewinkeltes Fe-C-O-Fragment:

Im Vergleich mit dem oxyMb-Zentrum ist das distale His nun nach rechts gedrängt, die Ausrichtung einer N-H-Funktion auf CO ist nicht gegeben. Lange Zeit wurden die beiden letzten Bilder so interpretiert, dass CO das distale His unter nennenswertem Energieaufwand zur Seite schiebt, woraus eine verminderte Bindungskonstante resultiert hätte. Aktuelle Untersuchungen haben diese Vorstellung nicht bestätigt. Das terminale O-Atom von O2 scheint im Vergleich mit dem O-Atom des Kohlenmonoxids vielmehr der deutlich bessere H-Brückenbindungs-Akzeptor zu sein. Die Bindung zu CO ist also nicht durch sterische Belastung besonders destabilisiert, die Bindung zu O2 ist vielmehr besonders stabil. Dieses Ergebnis legt nahe, dass die Natur eine höhere negative Beladung des terminalen Atoms des O2-Liganden gegenüber dem CO-Sauerstoffatom für eine starke H-Brückenbindung nutzt. Diese Schlussfolgerung leitet zu einem Vergleich der Bindungsverhältnisse in oxyMb und MbCO über, die eine Gemeinsamkeit verbindet: beide Komplexe liegen im Singulett-Grundzustand vor.
Versuch 28-15: Oxidation von Kupfer(I) durch Luftsauerstoff.
Teilschritte wie
2 CuII + O22− → 2 CuI + O2
oder
CuII + O2•− → CuI + O2
also die Reduktion von Kupfer(II) zur üblicherweise unbeständigen Oxidationsstufe +I durch geläufige Oxidationsmittel sind in der „normalen“ wässrigen Chemie undenkbar. In den Zentren von Hc und CuZnSOD finden diese Reaktionen statt. Die Voraussetzung ist das Zusammentreffen zweier Enzymcharakteristika: (1) die im vorangegangenen Kapitel eingeführte Reaktivitätssteuerung durch die Stabilisierung von Grenzorbitalen und (2) die Steuerung der Elektronenbilanz.
Hämocyanin ist das Sauerstofftransportmolekül von Schnecken und Tintenfischen („Mollusken“) sowie Krebsen, Skorpionen und Spinnen („Arthropoden“). Hc-Einzelmoleküle bilden artspezifisch Oligomere, deren Sinn im Aufbau von Kooperativität des O2-Transports zu liegen scheint (vgl. Mb und Hb). Neben Myoglobin/Hämoglobin und Hämerythrin ist Hämocyanin der dritte O2-Transporter, der in Organismen gefunden wird.
Hämocyanin ist ein Typ-3-Kupferprotein. Charakteristisch für diese Enzymklasse sind zweikernige Kupferzentren, die zur Aktivierung von O2 in der Lage sind – im Sinne von O2-Transport, als Oxygenase oder als Oxidase.
Es liegen Röntgenstrukturanalysen der oxygenierten und der deoxygenierten Form des Hämocyanins von Limulus polyphemus, des Pfeilschwanzkrebses, vor. Das Bild zeigt die Struktur des mit 628 Aminosäuren recht großen Proteins in einer Auflösung von 2.4 Å):

In dem mit Sauerstoff beladenenen aktiven Zentrum beträgt der Abstand der Kupferatome in dem durch einen Peroxo-Liganden verbrückten CuII2-Paar 3.6 Å:

Im deoxygenierten CuI2-Zentrum von Pfeilschwanzkrebs-Hämocyanin ist der Cu-Cu-Abstand mit 4.6 Å deutlich größer:

Die Abgabe von Sauerstoff unter Reduktion von Kupfer(II) zu Kupfer(I) steht in krassem Widerspruch zur Laborerfahrung und zur Lage der elektrochemischen Potentiale. Farblose Amminkupfer(I)-Lösungen werden durch Luftsauerstoff schnell und irreversibel zu den bekannten blauen Amminkupfer(II)-Lösungen oxidiert. Dass bei Hc Sauerstoff unter Kupfer(I)-Bildung freigesetzt wird, liegt am Zusammentreffen zweier enzymtypischer Faktoren.
(1) Auch in Hc fehlt eine regulär quadratische Koordination von Liganden, also die Anordnung, die ein stark destabilisiertes x2−y2-Orbital erzeugt und als Folge ein niedriges Potential. Beim Redoxpaar [CuI/II(NH3)4]+/2+ ist genau dies der Fall. Das Potential des Amminkupfer-Paares ist daher mit ca. 0 V kleiner als das Hc-Potential von ca. 0.3–0.4 V.
(2) Ein isoliertes binucleares Zentrum mit den zugänglichen Oxidationszuständen MI2, MIMII und MII2 erlaubt den Umsatz von maximal zwei Elektronen. Die mit höherer Triebkraft verlaufende 4-Elektronen-Reduktion von O2 (E0' = 0.82 V; siehe Anhang II) kann in einem solchen Zentrum nicht ablaufen. Möglich ist bestenfalls der 2-Elektronenprozess Sauerstoff/Peroxid – dessen Potential sich perfekt mit dem Hc-Potential deckt (Anhang II).
Die verwandte und thermodynamisch ebenfalls unmögliche Reaktion
CuII + O2•− → CuI + O2
läuft unter analogen Randbedingungen in Kupfer-Zink-Superoxiddismutase (CuZnSOD) ab:
Die Chemie von Hämocyanin und CuZnSOD ist durch die Festlegung der Elektronenbilanz aufgrund der Zahl der Redoxzentren und deren verfügbarer Oxidationsstufen festgelegt. Die Katalyse einer 2-Elektronen-Oxidation durch ein einkerniges Kupferprotein muss daher irritieren. Ist in Galactose-Oxidase Kupfer(III) in Betracht zu ziehen?
CcO ist das letzte Enzym der Atmungskette. CcO wird durch vier Äquivalente Cytochrom c reduziert, um anschließend ein Sauerstoffmolekül in einem Schritt zu Wasser zu reduzieren. Die gewonnene Energie wird als Protonengradient gespeichert. Die große Zahl von vier Elektronen für die Reaktion
O2 + 4 e− + 4 H+ → 2 H2O
stellt besondere Anforderungen an den Aufbau des aktiven Zentrums.
CcO besteht aus 26 Proteinketten mit insgesamt 3614 Aminosäuren. Es liegt eine Strukturanalyse von Rinderherz-CcO im oxidierten Zustand mit 1.8 Å Auflösung vor. Das O2-reduzierende Häm-a3-CuB-Zentrum kommt zusammen mit einem weiteren Häm-a in zwei Proteinketten vor. Abgebildet ist eine dieser beiden fast ausschließlich aus α-Helices bestehenden Ketten (A), zusätzlich ist im oberen Teil der Abbildung die an Faltblattabschnitten reichere Kette B. Zwischen A und B findet sich ein elektronenleitendes CuA-Zentrum:

Das abgebildete Protein reicht durch die innere Mitochondrienmembran. Der untere Teil ragt in das Innere der Organelle, der obere Teil ragt in den Transmembranraum zwischen innerer und äußerer Mitochondrienmembran. Die durch Cyt c herantransportierten Elektronen werden vom CuA-Zentrum angenommen und an das Häm-a-Zentrum weitergegeben. Der Elektronenfluss endet beim sauerstoffreduzierenden Häm-a3-CuB-Zentrum. Die vier Protonen der Bruttogleichung stammen aus dem Inneren des Mitochondriums (im Bild unten).
Beim Häm-a3-CuB-Zentrum fällt vor allem die Umgebung des Kupferatoms auf. Diese besteht aus drei His-Liganden, deren einer eine direkte Bindung zu einem Tyr-Rest aufweist:

Zur Reduktion eines O2-Moleküls wird dieses zwischen den beiden Metallzentren gebunden und in einem Schritt mit 4 Elektronen beladen.
Versuch 18-1: H2O2 entsteht bei der Verbrennung von Wasserstoff beim Abschrecken der Flamme
Versuch 3-16: Katalytische H2O2-Zersetzung durch Platin
Versuch 3-15: Katalytische H2O2-Zersetzung durch Kartoffel-Katalase
Versuch neu: Katalytische H2O2-Zersetzung durch Rinderserum-Katalase
Die Reaktivität eines Häm-Zentrums lässt sich durch die Wahl der zusätzlichen Liganden steuern. In Cytochrom c blockieren zwei weitere Liganden das Eisenatom für eine Substratbindung, so dass nur der Elektronenausstausch möglich wird. Bei der Häm-Katalase führt ein anionischer Ligand zur Stabilisierung höherer Oxidationsstufen als bei Mb und Hb möglich.
Häm-Katalase dient der Disproportionierung von H2O2, das zum Beispiel von SODs produziert wird:
H2O2 → H2O + ½ O2
Bei der Häm-Katalase ist die biologische Funktionseinheit wahrscheinlich das Tetramer der abgebildeten asymmetrischen Einheit:

Die Proteinstruktur hat wenig Ähnlichkeit mit Myoglobin und doch ist das aktive Zentrum eine verblüffend konservative Variation des Mb-Zentrums. Der Cofaktor des Apoproteins ist ein Häm b. Oberhalb des Häms befindet sich wie in Mb und Hb ein vor allem durch Phenylalanin-Seitenketten hydrophob ausgekleideter Raum, der auch ein „distales“ His enthält. In dessen Umgebung findet sich ein Wassermolekül in einem N···O-Abstand von 2.65 Å. 2.42 Å vom Wassermolekül entfernt ist ein O-Atom eines an Eisen(III)(?) gebundenen HO2−-Liganden. Der auffälligste Unterschied zu metMb ist der Ersatz des proximalen His durch einen Tyrosinato-Ligand, der die dreiwertige Stufe des Eisens als reduzierte Form des Redoxpaares FeIII/FeIVPor•+ stabilisiert.

Die H2O2-Disproportionierung erfordert einen 2-Elektronen-Redoxprozess. Im ersten Schritt wird die FeIII-Ruheform des Enzyms um zwei Elektronen oxidert, es entsteht eine Oxoferryl-Spezies, die anschließend wieder in die Ruheform zurückkehrt:
Por-FeIII + H2O2 → Por•+-FeIV=O + H2O
Por•+-FeIV=O + H2O2 → Por-FeIII + H2O + O2
Auf die Bedeutung der Superoxid-Beseitigung wurde bereits bei der MnSOD eingegangen. Im Vorkommen der einzelnen SODs spiegeln sich die stammesgeschichtlichen Zusammenhänge wider. FeSOD kommt in Prokaryonten vor. MnSOD, die sich wohl aus FeSOD entwickelt hat, kommt ebenfalls in Prokaryonten vor, aber auch in den Mitochondrien höherer Lebewesen. CuZnSOD schließlich – als jüngstes Enzym, in dem zwei Metalle kombiniert sind, die erst in einer schwefelfreien Sauerstoffwelt verfügbar wurden – findet sich im Cytosol aller Eukaryonten. Die Konzentration beträgt hier ca. 10−5 mol L−1. In [cuznsod2] wird errechnet, dass aufgrund der Konzentration und der Aktivität des Enzyms die Lebendauer von Superoxid-Radikalen um den Faktor 1010 gesenkt wird.
CuZnSOD gehört zu den Typ-2-Kupferproteinen, auch „nicht-blaue“ Kupferproteine genannt. Die UV/Vis-Spektren entsprechen im Wesentlichen denjenigen „normal“ koordinierter Kupfer(II)-Komplexe; der starke charge-transfer-Übergang der blauen Kupferproteine fehlt. Die weiter unten behandelte Galactose-Oxidase (GO) gehört ebenfalls zu den Typ-2-Zentren.
Die Struktur humaner CuZnSOD wurde mit einer Auflösung von 1.8 Å bestimmt:

Die Besonderheit des CuZnSOD-Zentrums ist ein zwischen Zink (links) und Kupfer (rechts) verbrückender Histidinato-Ligand. Der Abstand zwischen dem Kupfer(II)-Atom und dem verbrückenden NHis (gestrichelte Linie) beträgt ca. 2 Å in der oxidierten Form und mehr als 3 Å in der reduzierten. Die Strukturanalyse zeigt einen Mittelwert von ca. 2.6 Å, als dessen Ursache die Autoren teilweise Photoreduktion der oxidierten Ruheform während der Röntgenbestrahlung angeben:

Nach einem aktuellen Vorschlag in [cuznsod2] wird Superoxid auf dem Weg zum aktiven Zentrum protoniert und erreicht das Kupfer-Zentralatom als HO2 (pKA(HO2) = 4.8), wo es an die Stelle des Aqua-Liganden tritt (von anderen Autoren wird üblicherweise mit dem Superoxid-Anion formuliert und die beiden benötigten Protonen werden gesondert zugefügt). Das an Kupfer(II) in der Strukturanalyse gefundene Wassermolekül ist im folgenden Schema nicht berücksichtigt:

Die Nummerierung gilt für das in [cuznsod1] beschriebene humane Enzym. Oft untersucht ist Rinder-Erythrozyten-CuZnSOD, in der Kupfer von den His-Resten 44, 46, 61 und 118 koordiniert ist.
Der Katalysecyclus betont die Aussage, dass die Elektronendifferenz der verfügbaren Oxidationszustände die ablaufende Reaktion determiniert. Da Zink redox-inaktiv ist – seine Bedeutung wird in der Erhöhung der Acidität des verbrückenden His gesehen – sind ausschließlich 1-Elektronenschritte möglich. Genau dies verlangt die Superoxid-Disproportionierung.
Auch das elektrochemische Potential liegt im notwendigen Bereich. Zur Erinnerung: während das Potential eines Sauerstoff-Transporters dem Potential des genutzten Redoxprozesses entsprechen muss, so muss das Potential eines Disproportionierungskatalysators zwischen den Potentialen der beiden katalysierten Einzelschritte liegen.
Die folgende Graphik zeigt elektrochemische Potentiale bei pH 7 und Aktivität 1. Die Zahlenwerte sind entnommen: D. M. Kurtz jr.: Dioxygen-binding Proteins. CCC 8, 229–260 (230).
Die Zahl der übertragenen Elektronen ist farblich codiert:
schwarz: 1 Elektron,
blau: 2 Elektronen,
grün: 3 Elektronen,
rot: 4 Elektronen.
