 Das Liebig-Laboratorium
Lehramt AC1
Das Liebig-Laboratorium
Lehramt AC1
Verlängerte Anmeldung
Der Anmeldezeitraum für das Lehramt AC1 Grundpraktikum (LiebigLab, T1LB) wird bis einschließlich 17. Februar 2019 (23:59)
verlängert. Für eine nachträgliche Anmeldung schreiben
nutzen Sie bitte ausschließlich das Anmelde-Formular der
cup-Website:www.cup.lmu.de -> STUDIENGÄNGE -> Anmelden zu... -> Praktika -> AC Grundpraktikum Lehramt
https://www.cup.lmu.de/anmeld/acla1prak/index.php
Organisation
Das Liebig-Laboratorium (Lehramt AC1) ist ein gemeinsames Praktikum der Lehrbereiche Anorganische Chemie und Physikalische Chemie für das 1. Fachsemester im Lehramtsstudiengang Chemie und Biochemie. Die Laborräume befinden sich im 1. Stockwerk von Haus D.
Das Praktikum beginnt am 11. März 2019 und endet am 29. März 2019 und findet montags bis Freitags von 9:00–17:00 Uhr statt. Zu Beginn jedes Praktikumstages findet eine Vorbesprechung mit den Saalassistenten statt in der die am Praktikumstag durchzuführenden Versuche besprochen werden. Eine sinnvolle Versuchsdurchführung ist nur mit den entsprechenden Vorkenntnissen und der dazugehörigen Vorbereitung möglich. Alles, was an einem Tag gemacht wurde, bleibt am Platz stehen. Der Gruppenassistent geht ca. 1 Stunde vor Praktikumsende herum und prüft stichprobenartig, ob das praktisch Ausgeführte verstanden wurde (zum Beispiel Reaktionsgleichungen aufstellen lassen). Es herrscht während der gesamten Praktikumszeit Anwesenheitspflicht. Bei begründeter Abwesenheit (Attest, Nachweis) die sich über einen längeren Zeitraum hinzieht wird über die verpassten Praktikumsinhalte ein Kolloqium bei den jeweiligen Professoren abgehalten.
Hinweis: Studierende, die bei der Vorbesprechung nicht genügend vorbereitet sind, können durch die Saalassistenten für eine Stunde zur Nachbereitung versetzt werden. Falls dies mehr als zwei Mal auftritt gilt das gesamte Praktikum als nicht bestanden.
Die Vorbesprechung/Einführung inklusive Sicherheitseinweisung
zum Praktikum findet am 11.03.2019 von 9-11 Uhr
im Willstätter-Hörsaal statt. Die Teilnahme ist verpflichtend!
Eine der beiden folgenden Zulassungsvoraussetzungen muss am Termin der Vorbesprechung erfüllt sein, um das Praktikum antreten zu können:
- Bestandene Klausur zur Anorganische Chemie 1 (Experimentalvorlesung) (T1AA)
oder
- ACHTUNG NEUREGELUNG: Bestandene Klausur (mindestens 50% der maximalen Punktzahl) "Begleitvorlesung bzw. Seminar zum Chemischen Grundpraktikum" (T1LA) (Neuregelung durch den Prüfungsausschuss vom 18.05.2017)
Die Klausur (Allgemeine und Anorganische Chemie - AC 1) findet am Mo, den 11.02.2019, von 09:00-12:00 Uhr und 12:00-15:00 Uhr (in 2 Schichten) in allen CUP-Hörsälen statt.
Die Klausur (Begleitvorlesung zum Chemischen Grundpraktikum für Lehramtsstudierende) findet am Fr, den 15.02.2019, von 13:00-15:00 Uhr im Liebig- und Buchner-HS statt. (weitere Hinweise siehe Benotung weitere unten)
Die Wiederholungsklausur
(Begleitvorlesung
zum Chemischen Grundpraktikum für Lehramtsstudierende)
findet am Di, den 19.03.2019, von 13:00-15:00 Uhr im
Liebig-HS statt.
Die Ergebnisse
der Klausur (Begleitvorlesung
zum Chemischen Grundpraktikum für Lehramtsstudierende)
entnehmen Sie ab dem 19.02.2019 der Liste im Schaukasten im
Foyer von Haus E (neben dem Aufzug).
Eine Klausureinsicht zur Klausur (Begleitvorlesung zum Chemischen Grundpraktikum für Lehramtsstudierende) findet am 20.02.2019 von 13:00-14:00 Uhr in Raum E0.011 statt. Beachten Sie bitte, dass es nur einen regulären Termin für die Klausureinsicht gibt. Sollten Sie aus wichtigen, nicht verschiebbaren Gründen verhindert sein (Krankheit oder z.B.: amtliche, nicht verschiebbare Termine), so besteht die Möglichkeit, einen Sondertermin auszumachen, wenn Sie Ihre Verhinderung belegen können (ärztliches Attest, Amtliche Bescheinigung, etc.).
Die Ergebnisse
der Wiederholungsklausur (Begleitvorlesung
zum Chemischen Grundpraktikum für Lehramtsstudierende)
entnehmen Sie ab dem 21.03.2019 Mittag der Liste im Schaukasten im
Foyer von Haus E (neben dem Aufzug).
Eine Klausureinsicht zur Wiederholungsklausur (Begleitvorlesung zum Chemischen Grundpraktikum für Lehramtsstudierende) findet am 25.03.2019 von 8:00-9:00 Uhr in Raum E0.013 statt. Beachten Sie bitte, dass es nur einen regulären Termin für die Klausureinsicht gibt. Sollten Sie aus wichtigen, nicht verschiebbaren Gründen verhindert sein (Krankheit oder z.B.: amtliche, nicht verschiebbare Termine), so besteht die Möglichkeit, einen Sondertermin auszumachen, wenn Sie Ihre Verhinderung belegen können (ärztliches Attest, Amtliche Bescheinigung, etc.).
Weitere Informationen sind auf der Homepage von Prof. Dr. Thomas Bein einzusehen.
Zu diesem Praktikum ist ein Computereinführungskurs von 2 Std. verpflichtend. Die Kurse finden zwischen dem 22.-26.10.2018 statt.
Eine separate Anmeldung für den Computerkurs ist verpflichtend! Die Anmeldung finden Sie hier.
(Weitere Informationen erhalten Sie im CIP-Wiki der Fakultät Chemie/Pharmazie).
Bei weiteren Fragen wenden sie sich bitte an Daniel Böhm per E-Mail oder persönlich in Raum E3.002.
Das Einhalten der Sprechzeiten ist obligatorisch. Diese sind
Mo, Mi, Fr: von 10:00 bis 11:00 Uhr
E-Mail: Daniel.Boehm@cup.lmu.de
Benötigtes Arbeitswerkzeug ab dem 1. Praktikumstag
Alle
Erstsemester Studenten
erhalten am 1. Praktikumstag kostenlos einen neuen Laborkittel und eine
Schutzbrille, die Sie auch nach dem Praktikum behalten
dürfen
(und für weitere Praktika verwenden können).
Studenten aus höheren Semester müssen sich vor
Praktikumsbeginn eigenständig um Ihre
Schutzausrüstung bemühen.
Um die Spinde der Praktikumsräume verschließen zu können, benötigen Sie ein eigenes Schloss. Die Bügel sollten eine Stärke von ~5 mm haben und nicht zu kurz sein (kein kleines Kofferschloss).
Um die Laborschränke abschließen zu können benötigen Sie ein weiteres Schloss. Auch hier sollten die Bügel eine Stärke von ~5 mm haben und nicht zu kurz sein (kein kleines Kofferschloss).
Weiterhin benötigen Sie ein Taschenrechner, ein Periodensystem der Elemente,
einen Folienstift
(zum Beschriften von Glasgeräten) und Schreibzeug sowie
einen Block
für Notizen/Rechnungen.
Zusätzlicher Sicherheitshinweis: Das Tragen von
Kontaktlinsen
ist
nicht empfehlenswert, da im Notfall eine Augenspülung bei
Verätzung erschwert wird. Schutzbrillen können,
abhängig
vom Modell, über einer Brille getragen werden.
Am ersten Praktikumstag erhalten Sie außerdem eine
ausgedruckte Fassung des Skriptes. Diese ist die offizielle Version
für die Versuchsdurchführung. Trotzdem haben wir Ihnen im
Folgenden eine digitale Version bereit gestellt.
Begleitende Vorlesung und Literatur
Vorlesungen
Der Inhalt der drei Projekte aus der Anorganischen Chemie wird in der Vorlesung von Prof. Sünkel behandelt. Der Inhalt der Projekte aus der Physikalischen Chemie wird in der Vorlesung von Prof. Hartschuh behandelt.
Literatur
Abkürzungen im Text im Stil Mor-489K28.9 beziehen sich auf Mortimers Chemie. Lesen Sie die Abkürzung so: „Mortimer, Seite 486, Kapitel 28.9“. Die Seitennummern beziehen sich auf die 9. Auflage:
C. E. Mortimer, U. Müller: Chemie. 9. Auflage, Thieme 2007. Den „Mortimer“ können Sie innerhalb des Münchner Hochschulnetzes auch als „E-Book“ lesen.
Physikalisch-chemische Inhalte finden Sie im „Atkins“:
P. W. Atkins, J. de Paula: Physikalische Chemie. 4. Auflage, Wiley-VCH 2006.
Laborsicherheit
Machen Sie sich mit allen Aspekten der Laborsicherheit vertraut, die im Abschnitt zum Grundkurs zusammengestellt sind.
Aufbau der Projekte
Zum Kennenlernen der Projekte dienen Vorversuche, bei denen kurz Ausführung und Beobachtung protokolliert werden. In einigen Fällen sollen Reaktionsgleichungen formuliert werden.
Oft schließen sich dann Übungsanalysen an, die zum Kennenlernen der Methoden einer nachfolgenden Vollanalyse dienen.
Vollanalysen betreffen meist Proben aus der realen Umwelt oder detailliertere Untersuchungen. Bei einigen Vollanalysen wird Ihr Ergebnis im Anschluss mit einem Sollwert, der durch eine automatische Analysemethode selbst bestimmt wird, verglichen.
Das Ergebnis wird in einem kurzen gemeinsamen Protokoll zusammengestellt und es wird kurz darüber berichtet. Am Schluss des Protokolls wird zusammengestellt, wie die erwähnten Stoffe und Phänomene auf Englisch heißen.
Insgesamt setzt sich das Praktikum aus sechs Projekten zusammen, welche im Folgenden beschrieben werden.
Projektübersicht
Hier erhalten Sie den vorläufigen Ablaufplan (Stand: 27.02.2019) des Praktikums und eine Projektübersicht. (Es gilt jedoch immer die aktuellste Version des Ablaufplans in der Print-Version des Skriptes und welche im Saal ausgehängt wird (Datum beachten!))
Das vollständige Skript zum Praktikum (Stand: 27.02.2019) können Sie hier als digitale Version downloaden. Am ersten Praktikumstag erhalten Sie eine gedruckte Version (schwarz/weiß).
1. Grundkurs
Der Grundkurs zum Liebig-Lab soll Sie (1) mit den handwerklichen Grundlagen der nachfolgenden Projekte vertraut machen, (2) das Führen eines Laborprotokolls lehren, (3) mit den charakteristischen Eigenschaften wichtiger Stoffgruppen bekannt machen, und (4) an das Aufstellen von Reaktionsgleichungen heranführen. Der „rote Faden“ des Grundkurses ist der sichere Umgang mit Gefahrstoffen.
2. Farbe
Woher kommen Farbeindrücke? Farbreaktionen und -eindrücke bestimmen das tägliche Leben und sind auch ein wesentlicher Bestandteil der Analytischen Chemie. Tatsächlich haben „Farben“ ganz verschiedene Ursachen. In diesem Projekt werden grundlegende Phänomene, die zu Farbeindrücken führen, unterschieden und Messmethoden vorgestellt.
3. Elektrochemie
Elektrochemie bezeichnet mehrere verschiedene Teilgebiete innerhalb der Chemie. Sie ist zum einen eine Synthesemethode als präparative Elektrochemie, Elektrolyse oder Elektrosynthese, zum anderen ist sie ein Teilgebiet der physikalischen Chemie, welches sich mit dem Zusammenhang zwischen elektrischen und chemischen Vorgängen befasst. Die technische Chemie kennt neben großtechnisch angewandten elektrochemischen Synthesemethoden noch die Batterie- und Brennstoffzellentechnik, sowie die Galvanotechnik.
4. Anorganische-Chemie
Sie haben im ersten Teil des Praktikums neben grundlegenden
Arbeitstechniken auch den sicheren Umgang mit Gefahrstoffen geübt
sowie die Bedeutung von Betriebsanweisungen kennengelernt. Dieses
Wissen, dass chemische Substanzen in der Regel Gefahrstoffe sind und
einen besonnenen Umgang mit ihnen benötigen, "dürfen" Sie
nicht nur, sondern müssen Sie jetzt auch umsetzen!
Die Praktikumsanleitung zum Anorganisch-chemischen Teil kann
dieser vorläufigen
pdf-Version
entnommen werden.
5. Reaktionskinetik
Die Explosion von Nitroglycerin verläuft in Sekundenbruchteilen. Die Umwandlung von Diamant in Graphit verläuft dagegen unmessbar langsam (findet jedoch statt). In diesem Projekt soll ein erster Einblick in die Welt der Reaktionskinetik gegeben werden, indem der abstrakte, mathematische Begriff der Reaktionsordnung an einfachen Beispielen veranschaulicht und greifbar wird.
Analysen und Laborjournal
Über alle Versuche wird ein Laborjournal geführt, das von den Assistenten korrigiert wird und während der gesamten Praktikumszeit im Labor bleiben muss. Für jeden Versuch müssen die Fragen aus dem Skript schriftlich beantwortet und eventuelle Analysen / Ergebnisse angegeben werden. Über die Versuche der PC werden pro Zweiergruppe und Projekt Protokolle abgefasst und online als pdf abgegeben. Eine genaue Besprechung der Form und Abgabe erfolgt im Praktikum durch die Assistenten.
Für die Auswertung einiger PC-Versuche wird ein Programm zur Darstellung von Graphen benötigt (QtiPlot, OriginLabs,...). Eine Anleitung zum kostenlosen Programm QtiPlot, welches auf den CIP-Rechnern zur Verfügung steht, gibt die benötigten Funktionen des Programms umfassend wieder. Anwendungskenntnisse für MS Office werden vorausgesetzt.
Hilfreiche Hinweise für die Auswertung und Protokollerstellung im PC-Teil:
Zeitplan für die Protokollabgabe
Die Fragen zum Grundkurs
müssen im Laborjournal
bis zum 14.03.2019 (17.00 Uhr)
beantwortet werden.
Die Abgabe der Protokolle Farbe, Elektrochemie und Reaktionskinetik erfolgen per
E-Mail (von Ihrer cup-Mail Addresse) an
den Saal-Account an
den folgenden Tagen (immer jeweils bis 20:00 Uhr):
- Abgabe "Farbe" am 18.03.2019 (20.00Uhr)
- Abgabe "Elektrochemie" am 20.03.2019 (20.00Uhr)
- Abgabe "Reaktionskinetik" am 29.03.2019 (20.00Uhr)
Die Protokolle für den AC-Teil werden im Laborjournal angefertigt.
Zusätzlich müssen Wertetabellen ausgefüllt werden, die per
E-Mail an
den Saal-Account gesendet werden müssen (immer jeweils bis 20:00 Uhr):
Die Tabellen finden Sie hier.
- Abgabe "AC Tag1/2" am 21.02.2019
- Abgabe "AC Tag3/4" am 25.03.2019
- Abgabe "AC Tag5/6" am 27.03.2019
Verspätete Abgaben
werden mit je 0,3 Notenabzug pro
versäumten Tag berücksichtigt. Werden
Protokolle vollständig oder teilweise von anderen Quellen
abgeschrieben (beispielsweise Altprotokolle, unzitierte
Internetquellen) gilt das gesamte Protokoll als nicht bestanden und
muss erneut angefertigt werden.
Benotung
Die Vorlesung zum Praktikum und das Praktikum müssen beide separat bestanden werden. Zum Bestehen des praktischen Teils müssen alle Versuche einzeln bestanden werden.
Die Klausur zum Praktikum (Begleitvorlesung bzw.
Seminar zum Chemischen Grundpraktikum (T1LA)) findet am
Fr, den 15.02.2019,
von 13:00-15:00 Uhr im Liebig- und Buchner-HS
statt. Zur Zulassung für das
Praktikum müssen
mindestens 50% der
Punkte erreicht werden (Neuregelung durch den
Prüfungsausschuss
vom 18.05.2017). Die Klausur wird als bestanden gewertet bei erreichen von mindestens 50%
der Punkte. Die
Ergebnisse entnehmen Sie bitte der Liste (Aushang im Foyer Haus E
(neben dem Aufzug) ab dem 19.02.2019) - Es werden keine
Notenauskünfte per E-Mail erteilt! Die Möglichkeit zur
Klausureinsicht besteht am
20.02.2019 von 9:00-10:00 Uhr in Raum E0.013. Beachten
Sie bitte, dass es nur einen regulären Termin für die
Klausureinsicht gibt. Sollten Sie aus wichtigen, nicht verschiebbaren
Gründen verhindert sein (Krankheit oder z.B.: amtliche, nicht
verschiebbare Termine), so besteht die Möglichkeit, einen
Sondertermin auszumachen, wenn Sie Ihre Verhinderung belegen
können (ärztliches Attest, Amtliche Bescheinigung, etc.).
Die Wiederholungsklausur findet
am Di, den 19.03.2019, von 13:00-15:00 Uhr im Liebig-HS
statt.
Die Klausur wird als bestanden gewertet bei erreichen von mindestens 50%
der Punkte. Die
Ergebnisse entnehmen Sie bitte der Liste (Aushang im Foyer Haus E
(neben dem Aufzug) ab dem 22.03.2019) - Es werden keine
Notenauskünfte per E-Mail erteilt!
Die Möglichkeit zur
Klausureinsicht besteht am 25.03.2019 von 9:00-10:00 Uhr in Raum E0.013. Beachten
Sie bitte, dass es nur einen regulären Termin für die
Klausureinsicht gibt. Sollten Sie aus wichtigen, nicht verschiebbaren
Gründen verhindert sein (Krankheit oder z.B.: amtliche, nicht
verschiebbare Termine), so besteht die Möglichkeit, einen
Sondertermin auszumachen, wenn Sie Ihre Verhinderung belegen
können (ärztliches Attest, Amtliche Bescheinigung, etc.).
Beachten Sie, dass Sie erst zu den regulären Klausurterminen des darauffolgenden Wintersemesters die nächste Gelegenheit zur Klausurteilnahme haben, falls Sie auch die Wiederholungsklausur nicht bestehen.
Die Praktikumsnote setzt sich aus den folgenden Teilnoten zusammen: jeweils zur Hälfte aus der Praktikumsnote und der Klausurnote.
Technisches
Prüfen Sie hier, ob Ihr Browser das Skript korrekt darstellt.
… noch ein paar Tipps, wie Sie Fehler vermeiden können:
Diesen Kasten finden Sie am Ende mancher Projekte. Hier haben wir typische Fehler zusammengestellt, die uns in früheren Praktika aufgefallen sind. Am Besten lesen Sie diese Kommentare, bevor Sie die praktische Arbeit am Projekt beginnen.
Kurzanleitung zur Bedienung des UV/VIS Spektrometers
Das Ocean Optics Spektrometer ermöglicht die Messung von Absorptions- und Fluoreszenzspektren sowie die spektrale Charakterisierung unterschiedlichster Lichtquellen. Gesteuert wird das Spektrometer über ein NETBOOK und die Software SpectraSuite.
Aufbau der Messapparatur
Bauen Sie die Messapparatur wie in der Abbildung dargestellt auf:

Auf folgende Punkte sollten Sie beim Spektrometeraufbau achten:
• Auf keinen Fall die optische Faser knicken, oder in enge Radien biegen!
• Die Lichtquelle muss seperat eingeschalten werden und kann nur im SINGLE Modus betrieben werden.
• Sollte nach dem Programmstart das Spektrometer nicht erkannt worden sein, schließen Sie das Programm, trennen Sie die USB Verbindung zum Spektrometer, schließen dieses erneut an und starten Sie das Programm neu.
Nach dem Verbinden des USB-Kabels mit dem Netbook starten Sie das Programm „SpectraSuite“ vom Desktop aus und Sie erhalten folgenden Startbildschirm:
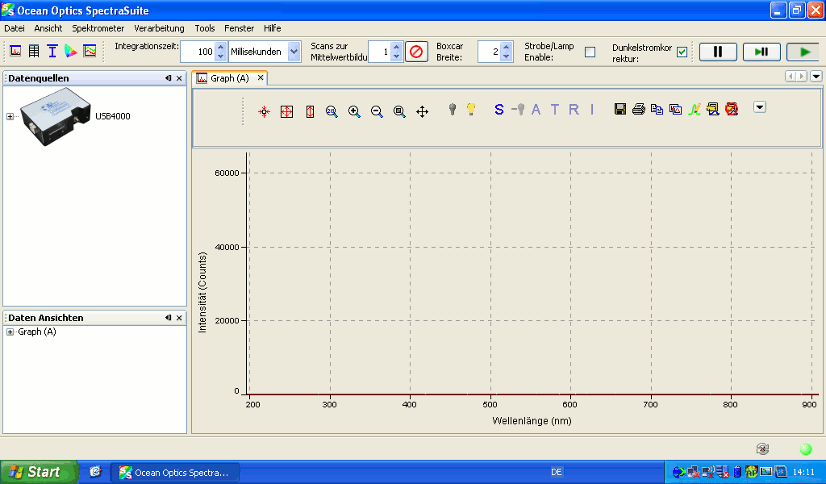
Beachten Sie, dass das Spektrometer bereits fortwährend Daten aufnimmt (erkennbar an der gedrückten Play-Taste rechts oben). Folgende in der Menüleiste enthaltene Buttons sind für die Messungen relevant:

- Play: Kontinuierliche Aufnahme von Spektren (
 )
)
- Pause: Stoppt die Aufnahme. Das letzte Spektrum wird angezeigt (
 )
)
- Play/Pause: Aufnahme eines einzelnen Spektrums (
 )
)
- Strobe/Lamp Enable: An- und Ausschalten der Lichtquelle
- Dunkelstromkorrektur: Option zur Verbesserung der Messung (immer angewählt lassen)

- Integrationszeit: Zeit, in der der Detektor Photonen für ein Spektrum zählt
- Scans zur Mittelwertbildung: Anzahl der Spektren, über die gemittelt wird
- Boxcar Breite: Anzahl der Pixel, über die gemittelt wird
Aufnahme von Spektren
Unabhängig vom eingestellten Aufnahmemodus, wird die Aufnahme der Spektren über die drei Schalter in der Menüleiste gesteuert (Play, Play/Pause, Pause). Eine sinnvolle Herangehensweise ist es, zunächst die kontinuierliche Aufnahme zu starten und sobald das gewünschte Ergebnis erreicht ist, das Spektrum durch Drücken des Schalters Pause festzuhalten. Über die verschiedenen Zoomwerkzeuge
 kann ein gewünschter Ausschnitt herausgestellt werden. Anschließend kann es, wie im Abschnitt Speichern von Spektren beschrieben, gespeichert werden.
kann ein gewünschter Ausschnitt herausgestellt werden. Anschließend kann es, wie im Abschnitt Speichern von Spektren beschrieben, gespeichert werden.
Aufnahme von Emissionsspektren

Um Emissionsspektren aufzunehmen, muss die Faser wie im Bild dargestellt mit dem Probenhalter verbunden werden. Ein Neutraldichte-Filter muss nicht eingesetzt werden. Verwenden Sie für die Messung eine 4-Wege-Küvette.
Schließen Sie alle geöffneten Graphen-Tabs und starten Sie eine neue Messung über Datei -> Neu -> Spektrumgraph. Das Programm nimmt nun Spektren im Scope-Modus auf [y-Achsenwert: Intensität (Counts)]. Zur Aufnahme eines Emissionsspektrums eignen sich folgende Einstellungen, die während der gesamten Messreihe unverändert bleiben müssen:
- Integrationszeit: 3,8 ms
- Scans zur Mittelwertbildung: 100
- Boxcar Breite: 2
Zur Verbesserung der Messung wird eine Dunkelkorrektur durchgeführt. Dazu wird ein einzelnes Spektrum der Probe bei ausgeschalteter Lichtquelle aufgenommen (Play/Pause). über den Button
![]() wird dieses Spektrum als Dunkelspektrum festgesetzt. Durch Anklicken des Buttons
wird dieses Spektrum als Dunkelspektrum festgesetzt. Durch Anklicken des Buttons
![]() wird der Scope-Modus mit ständigem Abzug des Dunkelspektrums aktiviert. Jetzt können Emissions-/Fluoreszenzspektren aufgenommen werden. Um die Dunkelkorrektur abzustellen, gelangt man über den Button
wird der Scope-Modus mit ständigem Abzug des Dunkelspektrums aktiviert. Jetzt können Emissions-/Fluoreszenzspektren aufgenommen werden. Um die Dunkelkorrektur abzustellen, gelangt man über den Button
![]() in dem normalen Scope-Modus zurück.
in dem normalen Scope-Modus zurück.
Aufnahme von Absorptionsspektren

Um Absorptionsspektren aufzunehmen, muss die Faser wie im Bild dargestellt mit dem Probenhalter verbunden werden und ein Neutraldichtefilter (ND-Filter), wie abgebildet, in den Strahlengang gesetzt werden. Verwenden Sie für die Messung eine 2-Wege-Küvette.
Zunächst werden, wie bei der Aufnahme von Emissionsspektren beschrieben, vorhandene Graphen-Tabs geschlossen und ein neuer geöffnet. Folgende Messparameter werden gewählt:
- Integrationszeit: 10 ms
- Scans zur Mittelwertbildung: 100
- Boxcar Breite: 2
Füllen Sie die Küvette mit dem im Versuch verwendeten Lösungsmittel, jedoch ohne die zu messende Probe. Zur Aufnahme eines Dunkelspektrums wird ein einzelnes Spektrum der Probe bei ausgeschalteter Lichtquelle aufgenommen (Play/Pause). Über den Button
![]() wird dieses Spektrum als Dunkelspektrum festgesetzt. Anschließend wird ein Lampenspektrum bei eingeschalteter Lichtquelle
wird dieses Spektrum als Dunkelspektrum festgesetzt. Anschließend wird ein Lampenspektrum bei eingeschalteter Lichtquelle
 aufgenommen und durch Anklicken des Buttons
aufgenommen und durch Anklicken des Buttons
![]() als Referenzspektrum (I0 -> siehe Vorlesungsskript Prof. Hartschuh) festgelegt. Jetzt kann auf Absorptionsmessung umgeschaltet werden
als Referenzspektrum (I0 -> siehe Vorlesungsskript Prof. Hartschuh) festgelegt. Jetzt kann auf Absorptionsmessung umgeschaltet werden
![]() und die Einheit der y-Achse wechselt automatisch auf „Absorption (OD)“.
und die Einheit der y-Achse wechselt automatisch auf „Absorption (OD)“.
Aufnahme von Transmissionsspektren
Zur Aufnahme von Transmissionsspektren werden die gleichen Einstellungen und Referenzspektren wie bei Absorptionsspektren verwendet. Lediglich der Messmodus wird auf Transmission umgeschaltet
![]()
Speichern von Spektren
Ist ein gewünschtes Spektrum festgehalten, kann es durch den Schalter
![]() oberhalb des Spektrums gespeichert werden. Im sich öffnenden Dialog
oberhalb des Spektrums gespeichert werden. Im sich öffnenden Dialog
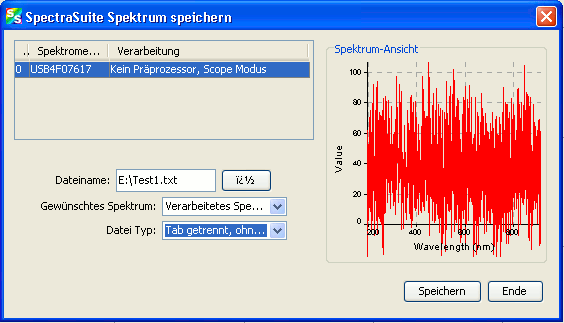
wird der Speicherort und der Dateiname angegeben. Wichtig ist die Einstellung des Datei-Typs „Tab getrennt, ohne Kopfzeile“, sonst kann das Spektrum anschließend von keinem externen Programm geöffnet werden.
Trend-Diagramm für Kinetiken
Ist bereits eine Absorptions- oder Fluoreszenzmessung wie vorher beschrieben konfiguriert, lässt sich die Änderung
in den Spektren auch zeitlich verfolgen und kann durch den Schalter
![]() oberhalb des Spektrums gespeichert werden. Dies wird dazu verwendet, um z.B. die Änderung der Optischen Dichte am Absorptionsmaximum im zeitlichen Verlauf einer Entfärbungsreaktion zu beobachten.
oberhalb des Spektrums gespeichert werden. Dies wird dazu verwendet, um z.B. die Änderung der Optischen Dichte am Absorptionsmaximum im zeitlichen Verlauf einer Entfärbungsreaktion zu beobachten.
Im sich öffnenden Dialog
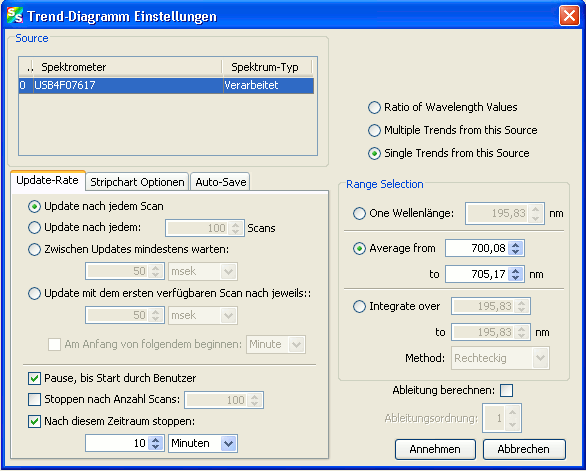
sind die Einstellungen wie abgebildet zu treffen. Lediglich die Angabe des Zeitraumes und der Bereichsauswahl sind abhängig vom jeweiligen Experiment. In der Bereichsauswahl wird der Bereich im Spektrum festgelegt, dessen zeitliche Änderung beobachtet werden soll (meist ist das der Bereich eines Absorptions- oder Emissionsmaximums). Die angegebenen Zahlen hier sind nur Beispiele!
Besonders wichtig ist auch noch die Eingabe der Auto-Save Optionen:
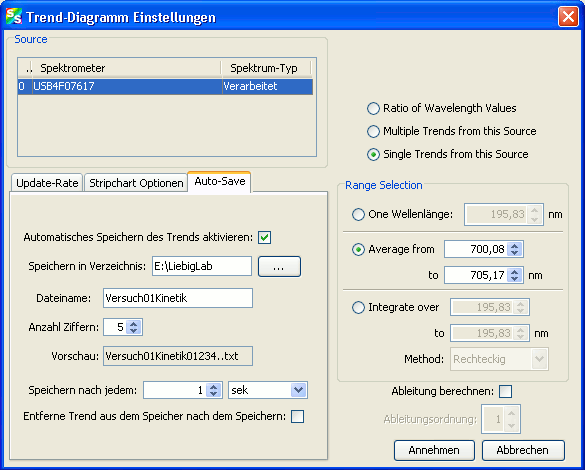
Aktivieren Sie das automatische Speichern, geben Sie den Speicherort und den Dateinamen an und geben Sie an, dass nach jeder Sekunde gespeichert werden soll. Nachdem alle Einstellungen getroffen wurden bestätigen Sie mit „Annehmen“. Darauf öffnet sich im Hintergrund ein weiterer Plot, in dem die Daten des Trend-Diagramms enthalten sind. Das im Vordergrund geöffnete Fenster kann geschlossen werden. Sollte eine Veränderung der Einstellungen nötig sein lässt sich diese Fenster jederzeit wieder über den Schalter „Konfigurieren“ aufrufen.
Sobald nun die Messung der Spektren im Hintergrund durch den Schalter
gestartet wurde, lässt sich die Aufnahme des Trends durch den kleinen Play-Schalter starten. Die dargestellten Daten werden automatisch im Hintergrund in die von Ihnen vorher benannte Datei gespeichert. Die Aufnahme des Trends kann durch den Schalter
![]() gestoppt werden und durch den Schalter
gestoppt werden und durch den Schalter
![]() zurückgesetzt werden, um einen weiteren Trend mit der fortlaufenden Nummerierung am Dateiende aufzunehmen. Alle Trenddiagramme werden automatisch im Hintergrund mit fortlaufender Nummerierung und unter dem vorher angegebenen Dateinamen gespeichert.
zurückgesetzt werden, um einen weiteren Trend mit der fortlaufenden Nummerierung am Dateiende aufzunehmen. Alle Trenddiagramme werden automatisch im Hintergrund mit fortlaufender Nummerierung und unter dem vorher angegebenen Dateinamen gespeichert.
FAQ
Manchmal erkennt der Rechner das Spektrometer nicht. Schritte zur Lösung dieses Problems:
• Programm neustarten
• Alle Anschlüsse überprüfen, USB-Kabel an beiden Anschlüssen ein- und ausstecken
• Rechner neustarten
Es sind keine vernünftigen Spektren zu erkennen:
• OD-Filter bei Absorption eingebaut? bzw. bei Fluoreszens ausgebaut
• Lampe angeschaltet/angesteckt?
• Küvette steht nicht gerade im Küvettenhalter?
Absorptionsspektren zeigen keinen sinvolle Absorptionskurve mit einem eindeutigen Maximum:
• Konzentration der betrachteten Farbstofflösung ist zu hoch. (Absorptionsmaximum liegt über > OD 1,5)
Grundkurs
Laborsicherheit
Betriebsanweisungen bilden den sicherheitstechnischen Rahmen des Praktikums. Dies beginnt für die Arbeit im Grundkurs und im Liebig-Laboratorium mit einer übergeordneten Anweisung.
Diese Allgemeine Betriebsanweisung fasst die Sicherheitsregeln des Praktikums allgemein und knapp zusammen. Sie basiert auf zwei Dokumenten, die zu Beginn der Anweisung als Quellen genannt sind: der GUV-R 120 Laboratorien und der Laboratoriumsordnung des Departments.
Als anschauliche Einführung liegt eine Broschüre der Gesetzlichen Unfallversicherung vor, der unter der Bestellnummer GUV-I 8553 herausgegebenen Einführung für Studierende mit dem Titel Sicheres Arbeiten in chemischen Laboratorien.
Sie erhalten zu Beginn des Praktikums jeweils ein Exemplar der Texte. Die dort angegebenen Regeln sind für Sie bindend.
Versuchsanleitungen
Das erste Hauptziel des Grundkurses ist es, mit Gefahrstoffen sicher umzugehen und soll durch Versuche erreicht werden bei denen die Warnungen der Betriebsanweisungen hinterfragt und verstanden werden. Behandelt werden die oben genannten wichtigsten Gefahrstoffe des ersten Semesters: Salz-, Schwefel- und Salpetersäure, Ammoniaklösung, Natron- und Kalilauge sowie Wasserstoffperoxid. Hinzu kommen wichtige Laborgase. Anschließend folgt das zweite Hauptziel des Grundkurses, nämlich eine Einführung in die Maßanalyse, die im Mittelpunkt des nachfolgenden Liebig-Laboratoriums stehen wird.
Handwerkszeug für die Laborarbeit
Arbeiten am Abzug
Das Arbeiten mit gefährlichen Stoffen, vor allem mit giftigen oder brennbaren Gasen und Dämpfen, ist nur in einem gut ziehenden Abzug erlaubt. Die Abzugsleistung ist im Wesentlichen davon abhängig wie weit der Frontschieber des Abzugs geöffnet ist. Bei weit geöffnetem Frontschieber verringert sich die Wirksamkeit des Abzuges erheblich. Gehen Personen am geöffneten Abzug vorbei, kann es zu Verwirbelungen und in deren Folge zum Ausbruch von Stoffen aus dem Abzug kommen. Halten Sie daher die Frontscheibe immer so weit wie möglich geschlossen und benutzen Sie zum Arbeiten nach Möglichkeit die seitlich verschiebbaren Arbeitsfenster. Neben dem Abführen von Gasen und Dämpfen dient der Abzug auch als Schutz vor verspritzenden Substanzen und als Splitterschutz. Trotzdem haben Sie, wie bei allen Arbeiten im Labor, auch beim Arbeiten am Abzug eine Schutzbrille zu tragen.
Wird am Abzug nicht gearbeitet, sind sowohl der Frontschieber als auch die Arbeitsfenster immer geschlossen zu halten! Der Abzug darf nicht als Lagerplatz verwendet werden. Entfernen Sie daher nicht benötigte Chemikalien oder Geräte aus dem Abzug und stellen Sie diese an die dafür vorgesehenen Lagerplätze. Schauen Sie nun, wie sich diese Erfahrungen in einer gerätebezogenen Betriebsanweisung niedergeschlagen haben.
Bunsenbrenner
Zum Erhitzen wird im Labor gewöhnlich der Bunsenbrenner benutzt. Im unteren Teil enthält er eine Düse, aus der Erdgas (Methan) ausströmt, und eine Vorrichtung, um Luft in verschiedenen Mengen in das Brennerrohr einzulassen. Ohne Luftzufuhr erhält man eine leuchtende Flamme. Ein Teil des Methans wird zunächst nur zu Kohlenstoff und Wasser oxidiert; bei der Flammentemperatur leuchten die gebildeten festen Kohlenstoff-(Ruß-)Teilchen. Leicht Sauerstoff abgebende Substanzen werden in der leuchtenden Flamme reduziert.
Bei Luftzutritt verbrennt Erdgas vollständig zu Kohlendioxid und Wasser. Bei der entstehenden „nichtleuchtenden“ Flamme lässt sich ein innerer, blau leuchtender Kegel erkennen, in dem reduzierende Bedingungen herrschen. Um diesen Kegel herum liegen oxidierende Bedingungen vor. An der Spitze des blauen Kegels ist die heißeste Stelle der Flamme:

Ist die Luftzufuhr zu groß oder der Gasdruck zu klein, so schlägt der Brenner durch, dass heißt, das Gas brennt im Innern des Brennrohres an der Gasaustrittsdüse. In diesem Fall muss die Gaszufuhr sofort abgestellt werden. Nach dem Erkalten des Brenners stellt man dann die Luftzufuhr etwas kleiner oder vergrößert die Gaszufuhr.
Analytische Waage

Waagen müssen stets sauber gehalten und schonend behandelt werden. Zunächst wird ein Wägegefäß auf die Waage gestellt (niemals Substanzen direkt auf die Waage geben!), dann wird „Tara“ gedrückt, um das Gefäß nicht mitzuwiegen. Nach dem Tarieren wird die zu wiegende Substanz abgewogen. Als Wägegefäß für Feststoffe dient ein Uhrglas, Wägeschiffchen oder ein anderes leichtes(!) Glasgefäß. Im Falle einer hygroskopischen, flüssigen oder leicht flüchtigen Substanz ist ein Gefäß mit eingeschliffenem Deckel zu verwenden. Nun kann die Wägung vorgenommen werden. Informieren Sie sich vor Beginn der Wägung anhand der Anleitung über die Bedienung der Waage.
Finden Sie mit Hilfe der Bedienungsanleitung heraus, warum Sie nicht irgend ein beliebig schweres Wägegefäß verwenden dürfen – schließlich tariert man doch.
Glasbearbeitung
Im Labor wird überwiegend mit Glasgeräten gearbeitet, weshalb Sie die einfachsten Arten der Glasbearbeitung beherrschen sollten, auch um kleinere Beschädigungen schnell zu beheben.
Schneiden von Glasrohren
Das Rohr wird mit einem scharfen Glasmesser zu einem Viertel seines Umfanges an der gewünschten Stelle eingeritzt. Durch leichtes Ziehen und Biegen gegen die Kerbe wird das Glasrohr auseinandergesprengt.
Rundschmelzen von Glasrohren
Zerteilte und abgesprengte Glasrohre oder Stäbe sind an den Schnittkanten scharf. Ihre Enden müssen daher rundgeschmolzen werden. In der leuchtenden Flamme wird das Rohrende unter gleichmäßiger Drehung erwärmt, anschließend wird bei entleuchteter Flamme erhitzt, bis die Kanten erweichen. Je weiter das Rohr ist, desto vorsichtiger muss die Erwärmung vor sich gehen. Zum Schluss wird in der leuchtenden Flamme getempert.
Glasrohr biegen
Um ein Rohr zu biegen, wird eine Seite mit einem Stopfen verschlossen. Anschließend wird unter gleichmäßigem(!) Drehen bis zum Erweichen des Glases erhitzt (wie oben mit leuchtender Flamme beginnen). Die erweichte Stelle wird nach unten weggebogen und eine eventuell entstandene Verengung außerhalb der Flamme vorsichtig wieder aufgeblasen. Zum Schluss wird in der leuchtenden Flamme getempert.
Reinigen von Laborgerät
Viele analytische Nachweisreaktionen sind sehr empfindlich, sie sprechen auf kleinste Substanzmengen an. Das bringt es mit sich, dass nachlässig gereinigtes Laborgerät zur Fehlinterpretation von Versuchen führt. Der folgende Versuch regt Sie dazu an, einen Spülvorgang als mehrfaches Verdünnen um einen charakteristischen Faktor zu begreifen.
Spülen als Verdünnungsreihe
Dieser Versuch zeigt sehr schön, dass ein Laborgefäß nach dem Ausspülen „sauber“ aussieht, es aber nicht immer ist.
In drei Reagenzgläser werden je 2 mL einer Eisen(III)-chloridlösung (ca. 0,1 mol L−1) gegeben. Nach dem Ausgießen der Lösung spült man das erste Glas mit 1 mL Wasser aus. Das zweite Glas wird zweimal mit je 1 mL Wasser ausgespült. Das dritte Glas wird unter Verwendung von Reagenzglasbürste und Spülmittel/Wasser gründlich gereinigt und anschließend mehrmals mit destilliertem Wasser ausgespült. Durch Zusatz von Ammoniumthiocyanat-Lösung (ca. 0,1 mol L−1) lassen sich noch vorhandene Eisen(III)-Ionen durch eine Rotfärbung nachweisen. Dies sollte bei den ersten beiden Reagenzgläsern der Fall sein, jedoch nicht bei dem sorgfältig gereinigten Glas.
Gehen Sie davon aus, dass beim Ausgießen immer 1 Tropfen im Glas zurückbleibt. Das Volumen eines Wassertropfens ist ungefähr 0,05 mL. Berechnen Sie die Eisen(III)-Konzentration in mol L−1 nach dem ersten und nach dem zweiten Wiederauffüllen mit je 1 mL Wasser.
Wichtige Handgriffe
Reagenzgläser werden für chemische Reaktionen und zur Aufbewahrung von kleinen Flüssigkeitsmengen verwendet. Zur Lagerung oder zum Durchmischen werden sie mit einem Kork- oder Kunststoffstopfen verschlossen (niemals die Öffnung mit dem Daumen verschließen). Gläser, die in einer Flamme erwärmt werden sollen, sind meist dünnwandig, um Bruch durch thermische Spannungen zu vermeiden.
Erhitzen im Reagenzglas
Einmal richtig und einmal falsch. Wenn Sie beides gesehen haben, werden Sie es nicht mehr falsch machen. Ziel ist, dass Sie einen Siedeverzug kennenlernen und eine Möglichkeit, diesen zu vermeiden.
Ein Reagenzglas wird etwa zur Hälfte mit Wasser gefüllt und mit Hilfe einer Reagenzglasklammer senkrecht in die nichtleuchtende Bunsenbrennerflamme gehalten. Innerhalb kurzer Zeit errreicht das Wasser Siedetemperatur. Es kommt in der Regel zu einem Siedeverzug, der den Inhalt des Reagenzglases herausschießen lässt; Reagenzglas nicht auf den Nachbarn richten!
Ein zweiter Versuch zeigt, wie ein solcher unerwünschter Siedeverzug vermieden wird. Das Reagenzglas wird nur etwa zu ¼ mit Wasser gefüllt. Dann wird es schräg in die nichtleuchtende Bunsenbrennerflamme gehalten und dabei geschüttelt. Das Wasser sollte nun gleichmäßig sieden und nicht wie vorher herausspritzen, da eine lokale Erhitzung vermieden wird.
Beschreiben Sie, wie es zu einem Siedeverzug kommen kann.
Mischen im Reagenzglas
Es geht um Fingerfertigkeit. Wenn Sie Mühe haben, kleine Substanzmengen so sicher zu dosieren, dass nichts überschäumt, wiederholen Sie den Versuch mehrmals.
In einem Reagenzglas wird ca. 1 mL gesättigte Natriumcarbonat-Lösung mit 2 Tropfen Methylrot-Indikatorlösung versetzt. Nun wird tropfenweise verdünnte Salzsäure hinzugegeben und durch Schütteln des Reagenzglases gemischt. Es wird weitere Säure zugegeben, bis der Indikator umschlägt.
Welches Gas entwickelt sich bei der Säurezugabe? Formulieren Sie eine Reaktionsgleichung.
Lösungen
Besonders wichtig: die üblichen Konzentrationsmaße. Die Stoffmengenkonzentration oder Molarität (c) wird definiert als Stoffmenge (n) pro Volumen (V), Einheit: mol L−1, abgekürzt m.
Der meist in Prozent angegebene Massenanteil („Massenprozent“) ist definiert als Masse pro Gesamtmasse des Gemisches, Einheit: 1, meistens %. 30%ige Salzsäure enthält daher 30 g HCl in 100 g Salzsäure.
Die Dichte von 30%iger Salzsäure ist 1,15 kg L−1. Wieviel mL dieser Säure müssen abgemessen werden, wenn 36,5 g HCl benötigt werden?
Fällung bei verschiedenen Konzentrationen
Die meisten Versuche werden in Lösung durchgeführt. Das Gelingen des Versuches hängt oft von der geeigneten Konzentration der Lösung ab. Arbeitet man mit zu konzentrierten Lösungen, so können Konzentrationsniederschläge auftreten oder aber die Niederschlagsmenge ist so groß, dass die charakteristische Form der Abscheidung (kristallin, flockig, gelartig) nicht oder nur schwer erkennbar ist. In zu verdünnten Lösungen kann eine Reaktion ganz ausbleiben (Grenzkonzentration, Erfassungsgrenze) oder mit großer zeitlicher Verzögerung eintreten.
Versetzen Sie eine kleine Probe einer 5%igen Eisen(III)-chloridlösung in einem Reagenzglas mit wenig konzentrierter Ammoniaklösung; eine zweite Probe der Lösung wird auf das 20-fache verdünnt und ein Teil der Probe mit konzentrierter Ammoniaklösung versetzt. Schließlich versuchen Sie, wie weit die Lösung verdünnt werden muss, damit mit Ammoniaklösung keine deutliche Fällung mehr auftritt.
Formulieren Sie die Gleichung für die Reaktion von Eisen(III)-Ionen mit Ammoniaklösung.
Konzentrationsreihe
Eine Vorübung zu Abschnitt 3:
Stellen Sie eine Konzentrationsreihe aus einer 10 m HCl mit folgenden Konzentrationen her: 0,1 m, 0,001 m, 0,00001 m. Bestimmen Sie anschließend mit Hilfe von pH-Papier den pH-Wert der Lösungen.
Die einfache Arrhenius-Theorie (Säuren zerfallen [„dissoziieren“] in H+-Ionen und Säurerest-Anionen) erlaubt Ihnen, das Ergebnis einzuordnen. Nehmen Sie an, dass jedes Molekül HCl dissoziiert. Der pH-Wert ist als negativer Zehnerlogarithmus der H+-Konzentration definiert. Rechnen Sie für die drei Konzentrationen die zu erwarteten pH-Werte aus und vergleichen Sie mit Ihrem Ergebnis.
Gemische
Unter einem Gemisch versteht man einen Stoff, der mindestens aus zwei Reinstoffen besteht. Die spezifischen Eigenschaften wie zum Beispiel Dichte, Siedepunkt oder Farbe sind vom Mischungsverhältnis (Massenverhältnis) der Komponenten abhängig. Man unterscheidet homogene Gemische, bei denen man weder mit dem Auge noch mit dem Mikroskop die Zusammensetzung verschiedener Reinstoffe erkennen kann und heterogene Gemische.
Quantitative Trennung eines Gemisches
Das zu trennende Dreikomponenten-Gemisch enthält Kupfer(II)-chlorid-Dihydrat, Zimtsäure und Sand. Löslichkeiten in Wasser: CuCl2·2H2O: bei 0 °C 1,10 g mL−1, bei 100 °C 1,92 g mL−1; Zimtsäure: bei 0 °C 0,0004 g mL−1, in der Siedehitze > 1,60 g mL−1 (wenn etwas Ethanol zugesetzt wird); SiO2: praktisch unlöslich.
Zunächst werden 5 g des Gemisches in ein kleines Becherglas gegeben, 10 mL Wasser hinzugefügt und 1 Minute gerührt. Es wird nun abfiltriert. Wenn der Rückstand im Filter fast trocken ist, wird mit ca. 1 mL Wasser gewaschen. Der beschriebene Waschvorgang wird solange wiederholt, bis das ablaufende Filtrat keinen Anteil der gut löslichen Komponente mehr zeigt. Das Filtrat stellt man zunächst (weiter bei 1) zur Seite (Welche Komponente befindet sich im Filtrat?).
Das Filterpapier mit dem Rückstand gibt man mit 20 mL Wasser und 10 mL Ethanol in einen Erlenmeyerkolben, wäscht es darin, entfernt das Papier und erhitzt auf der Heizplatte unter Rühren zum Sieden. Dann wird möglichst heiß durch einen erwärmten Trichter filtriert. Der Rückstand im Filter wird zweimal mit je 10 mL heißem Wasser gewaschen. Das Filtrat wird nun auf Zimmertemperatur abgekühlt und man stellt es mindestens 40 Minuten in ein Eiswasser-Bad (weiter bei 2).
Während der Wartezeit wird der Filterrückstand mit Wasser gewaschen und getrocknet. Um welche Komponente handelt es sich? Bestimmen Sie die Masse.
2 Das gebildete Kristallisat wird mit Hilfe einer Nutsche isoliert und gut trocken gesaugt. Die gewonnene Komponente wird zum weiteren Trocknen in den Exsikkator gegeben. Am nächsten Praktikumstag wird der Schmelzpunkt und die Masse bestimmt. Vergleichen Sie Ihren Wert für den Schmelzpunkt mit dem Literaturwert von 134–136 °C. Um welche Komponente handelt es sich?
1 Das farbige Filtrat der ersten Trennung wird unter starkem Rühren bis fast zur Trockene eingedampft (am Schluss vorsichtig und nicht zu stark erhitzen). Erscheint wasserfreies Kupfer(II)-chlorid (Farbe!), so beendet man das Heizen. Bestimmen Sie die Masse.
Gefahrstoffe
Die Arbeit mit Gefahrstoffen ist in einem chemischen Laboratorium Alltag. In diesem Kapitel steht daher ein Lernziel im Vordergrund: der sichere Umgang mit Gefahrstoffen. Die vom Gesetzgeber geforderte praktische Hilfe hierzu sind die Betriebsanweisungen. Die ersten Betriebsanweisungen hatten Sie oben bereits kennengelernt, nämlich die Allgemeine Betriebsanweisung, die den Rahmen für das sichere Arbeiten im Praktikum bildet und die gerätebezogene Betriebsanweisung für den Umgang mit Laborabzügen. In den stoffbezogenen Betriebsanweisungen dieses Kapitels sind oft Gefahren genannt, ohne dass jedoch deren chemischer Hintergrund immer beleuchtet wird. Es bleibt also die Aufgabe zu verstehen, was bei einer Reaktion geschieht und worin die Gefahr besteht. Sie werden daher solche gefährlichen Reaktionen untersuchen, den Stoffumsatz durch eine Reaktionsgleichung ausdrücken und die Ursache der Gefahr erkennen.
Sie erkennen Versuche, durch welche die Betriebsanweisungen näher beleuchtet werden, an der Gestaltung der Versuchsüberschrift in der (derzeit noch) orangen Warnfarbe und dem nachgestellten Vermerk „(BA!)“:
Warnung in der Betriebsanweisung
Die Betriebsanweisungen der in diesem Kapitel verwendeten Ausgangsstoffe sind bereits in diesem Skript verlinkt. Allerdings entstehen bei einigen dieser Versuche weitere Stoffe, von denen eine Gefahr ausgehen kann, beispielsweise giftige oder brennbare Gase. Machen Sie sich also vor der Durchführung der Versuche klar, welche Stoffe entstehen können (Reaktionsgleichung!) und informieren Sie sich gegebenfalls selbstständig anhand von Betriebsanweisungen über mögliche Gefahren. Hierzu steht ein Ordner mit Betriebsanweisungen im Praktikumssaal aus.
Salzsäure, HCl
Salzsäure gehört zu den wichtigsten Grundchemikalien des Labors. Im menschlichen Organismus kommt sie als „Magensäure“ in einer Maximalkonzentration von ca. 0,1 mol L−1 vor. Bereits diese Konzentration reicht aus, um bei Refluxerkrankungen („Sodbrennen“) die Speiseröhre zu verätzen. Die im Labor verwendete Salzsäure ist bis zu 100-mal höher konzentriert.
Eigenschaften
Konzentrierte Salzsäure ist eine ca. 35%ige Lösung des farblosen und stechend riechenden Gases Chlorwasserstoff (HCl) in Wasser, verdünnte Salzsäure ist eine ca. 7%ige Lösung. Wird konzentrierte Salzsäure erhitzt, so destilliert hauptsächlich Chlorwasserstoff und wenig Wasser ab bis die Konzentration der Lösung auf 20 % gesunken ist. Erhitzt man umgekehrt verdünnte Salzsäure, so entweicht hauptsächlich Wasser, bis die Säure wieder 20%ig ist. Die Dichte einer Salzsäure folgt einer einfachen Faustformel: Division der Prozentangabe durch 200, dann Addition von 1 ergibt die Dichte in g cm−3. 34%ige Salzsäure hat also wegen 34/200 + 1 = 1,17 die Dichte 1,17 g cm−3.
Errechnen Sie die Stoffmengenkonzentration (Molarität) von konzentrierter und verdünnter Salzsäure.
Salzsäure reagiert mit hinreichend starken Reduktionsmitteln heftig unter Wasserstoffentwicklung, während starke Oxidationsmittel Chlor freisetzen. Neben der großen Exothermie der Reaktionen ergeben sich besondere Gefahren aus der Brennbarkeit von Wasserstoff und der Aggressivität von Chlorgas.
Handhabung
Der Umgang mit konzentrierter Salzsäure (ab 25 %) wird durch eine Betriebsanweisung geregelt.
Reaktion von Aluminium mit Salzsäure
Die Betriebsanweisung zur konzentrierten Salzsäure führt deren Umsetzung mit Aluminium als besondere Gefahr auf. Der Versuch soll aufzeigen, wieso.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Führen Sie die Versuche, bei denen eine Gasentwicklungsapparatur benötigt wird, in Zweiergruppen aus.
Geben Sie ca. 250 mg Aluminium-Grieß in den 100-mL-Einhalsrundkolben, der Ihnen für diesen Versuch zur Verfügung gestellten Gasentwicklungsapparatur. Füllen Sie die beigegebene Kristallisierschale (hier als pneumatische Wanne zu verwenden) mit ausreichend Wasser. Legen Sie darin unter Wasser einen passenden Gummistopfen für ein Reagenzglas bereit, in dem das bei der Reaktion freigesetzte Gas gesammelt werden soll, und tauchen Sie das vollständig mit Wasser gefüllte Reagenzglas in das Wasser der pneumatischen Wanne ein. Versetzen Sie nun im Reaktionskolben das Aluminium mit ca. 10 mL verdünnter Salzsäure. Verschließen Sie rasch den Kolben mit dem Gasüberleitungsrohr, das in einem durchbohrten Stopfen steckt, und tauchen Sie dessen Ende unter die Wasseroberfläche der pneumatischen Wanne. Es setzt langsam Gasentwicklung ein, die nach kurzer Zeit heftiger wird. Beginnen Sie mit dem Auffangen des Gases erst nach ca. 2 Minuten, so dass zunächst die Luft aus dem Reaktionskolben verdrängt wird. Halten Sie dann das Ende des Gaseinleitungsrohres direkt unter das senkrecht aufgerichtete Reagenzglas. Wenn das Wasser im Reagenzglas vollständig verdrängt ist, wird es unter Wasser mit dem Gummistopfen fest verschlossen. Nehmen Sie nun das verschlossene Glas aus dem Wasser. Ziehen Sie dann den Stopfen ab und halten Sie die Öffnung des Reagenzglases sofort in die Flamme des Bunsenbrenners (Öffnung stets nach unten weisen lassen).
Um welches Gas handelt es sich (Reaktionsgleichung)? Warum führt die Betriebsanweisung die Reaktion mit Aluminium als Gefahr auf?
Reaktion von Kaliumpermanganat mit Salzsäure
Die Betriebsanweisung zur konzentrierten Salzsäure führt deren Umsetzung mit Kaliumpermanganat als besondere Gefahr auf. Der Versuch soll aufzeigen wieso.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Geben Sie ca. 800 mg gepulvertes Kaliumpermanganat in angehäufter Form auf ein Uhrglas. Tropfen Sie anschließend mittels einer Pasteurpipette langsam konzentrierte Salzsäure zu. Dabei entsteht ein Gas. Die Farbe des Gases können Sie deutlicher wahrnehmen, indem Sie das Experiment vor einem weißen Hintergrund (zum Beispiel einem Bogen weißen Papiers) durchführen.
Um welches Gas handelt es sich (Reaktionsgleichung)? Warum führt die Betriebsanweisung die Reaktion mit Kaliumpermanganat als Gefahr auf?
Wasserstoffentwicklung im Labor
Bei den Versuchen mit Salzsäure ergab sich die Entstehung von Wasserstoff als eine wesentliche Gefahrenquelle. Andererseits gehört Wasserstoff zu den wichtigsten Laborgasen überhaupt. Die sichere Herstellung und Handhabung von Wasserstoff gehört daher zu den Grundoperationen in einem chemischen Laboratorium.
Wasserstoff ist ein farbloses, brennbares und geruchloses Gas, das viel leichter als Luft ist. Es bildet mit Luft, mehr noch mit reinem Sauerstoff oder auch Chlor, explosionsfähige Gemische. Im Laboratorium wird Wasserstoff bei der Einwirkung von Säuren auf Zink oder von Laugen auf Aluminium erhalten, in der belebten Natur produzieren und nutzen Mikroorganismen Wasserstoff durch Hydrogenase-Enzyme. Während Wasserstoff in Wasser oder in Säuren die Oxidationsstufe +I aufweist, kann er mit Metallen Hydride mit der Oxidationsstufe −I bilden, wie beispielsweise in LiH.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Geben Sie 2 Zinkgranalien in den Rundkolben der für diesen Versuch bereit gestellten Gasentwicklungsapparatur und versetzen Sie diese mit ca. 5 mL konzentrierter Salzsäure. Fangen Sie das entstehende Gas wie zuvor beschrieben in einem Reagenzglas auf und führen Sie eine Knallgasprobe durch. Möchte man die Wasserstoffentwicklung grob steuern, kann man dies durch dosiertes Zutropfen der Salzsäure unter Verwendung eines Tropftrichters erreichen.
Wieviel Liter Wasserstoffgas lässt sich unter Standardbedingungen aus 100 g Zink und einer ausreichenden Säuremenge gewinnen?
Schwefelsäure, H2SO4
Schwefelsäure ist eine der wichtigsten Grundchemikalien. Die Schwefelsäureproduktion gehört zu den Kennzahlen, die in die Bewertung der Wirtschaftsleistung einer Volkswirtschaft eingehen. Im Gegensatz zu der aus zwei Dritteln Wasser bestehenden konzentrierten Salzsäure ist konzentrierte Schwefelsäure fast wasserfrei.
Eigenschaften
Konzentrierte Schwefelsäure, eine ölige Flüssigkeit, ist 96%ig. Die chemischen Eigenschaften sind durch zwei Charakteristika geprägt: Schwefelsäure ist (1) außerordentlich wasseranziehend und (2) stark oxidierend. „Wasseranziehend“ bezieht sich dabei unmittelbar auf den Stoff Wasser selbst, darüberhinausgehend aber auch auf komplexe chemische Reaktionen, in deren Verlauf das Reaktionsprodukt Wasser entsteht. Achten Sie hier besonders auf den Versuch mit Ameisensäure. Starke Reduktionsmittel wie Zink reagieren mit heißer konzentrierter Schwefelsäure unter sehr weit gehender Reduktion der Säure zu elementarem Schwefel und in geringem Umfang sogar zu Hydrogensulfid, H2S. Beachten Sie bei den Versuchen den Einfluss der Reaktivität des Metalls und der Konzentration der Säure.
Handhabung
Der Umgang mit Schwefelsäure (ab 5 %) wird durch eine Betriebsanweisung geregelt.
Bei der Herstellung von verdünnter Schwefelsäure wird stets die konzentrierte Säure langsam und unter guter Durchmischung in das Wasser gegossen – nicht umgekehrt: „erst das Wasser, dann die Säure, sonst geschieht das Ungeheure“! Heiße konzentrierte Schwefelsäure darf keinesfalls verdünnt oder in den Ausguss gegossen werden.
Verdünnen von konzentrierter Schwefelsäure
Nochmal, auf die Reihenfolge kommt es an: „Erst das Wasser, …“
Zu 3 mL Wasser gieße man aus einem zweiten Reagenzglas etwa den gleichen Raumteil konzentrierter Schwefelsäure. Die Mischung erwärmt sich stark.
Jetzt, wo Sie die freiwerdende Wärmemenge gespürt haben: Was ist denn das „Ungeheure“? Also was genau kann passieren, wenn Wasser in konzentrierte Schwefelsäure gegossen wird?
Reaktion mit Ameisensäure
Das Verkohlen organischen Materials bei Schwefelsäurezusatz ist formal ein Entzug der Elemente des Wassers. Ein Beispiel ist das Verkohlen von Traubenzucker:
C6H12O6 → 6 C + 6 H2O.
Ameisensäure ist einer der seltenen Fälle, bei denen bei der Umsetzung mit konzentrierter Schwefelsäure anstelle eines undefinierbaren Teers ein wohldefiniertes Reaktionsprodukt entsteht.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Versetzen Sie in einem Reagenzglas ca. 2 mL konzentrierte Ameisensäure mit ca. 1 mL konzentrierter Schwefelsäure. Sollte es bei Raumtemperatur noch nicht zu einer Gasentwicklung kommen, erwärmen Sie ein wenig.
Welches Gas entsteht bei der Reaktion? Formulieren Sie die Reaktionsgleichung. Geben Sie die Lewisformeln von Ameisensäure und des entstehenden Gases an.
Reaktion von Zink mit verdünnter Schwefelsäure
Verdünnte Schwefelsäure löst viele Metalle wie Eisen, Aluminium und Zink unter Wasserstoffentwicklung zu Sulfaten auf.
Man übergieße in einem Reagenzglas Zinkgranalien mit verdünnter Schwefelsäure, die zuvor mit einigen Tropfen konzentrierter Schwefelsäure versetzt wurde. Das Zink wird aufgelöst und Wasserstoff entweicht.
Formulieren Sie die Reaktionsgleichung. Welche Gefahr geht von dieser Reaktion aus?
Reaktion von Eisen mit konzentrierter Schwefelsäure
Konzentrierte Schwefelsäure verhält sich zum Beispiel gegenüber Eisen ganz anders als die verdünnte Säure. Sie löst das Metall bei Zimmertemperatur nicht auf. Bei höherer Temperatur bildet sich das Sulfat, aber es wird kein Wasserstoff frei, sondern es entwickelt sich Schwefeldioxid, SO2.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Geben Sie in einem Reagenzglas zu einigen Eisenspänen (Eisenpulver) ca. 3 mL konzentrierte Schwefelsäure und erwärmen Sie das Gemisch in der Bunsenbrennerflamme bis zur einsetzenden Gasentwicklung. Halten Sie in das entweichende Gas einen feuchten Universalindikatorstreifen.
Formulieren Sie die Reaktionsgleichung. Erklären Sie die Farbänderung des Indikatorpapiers. Welche Gefahr geht von dieser Reaktion aus?
Reaktion von Zink mit konzentrierter Schwefelsäure
Ein unmittelbarer Nachweis für das Vorhandensein von Schwefel in der Schwefelsäure: im oberen Teil des Reagenzglases bildet sich ein gelber Beschlag von festem Schwefel, und gelbe Schwefeltröpfchen scheiden sich ab. Entweichendes Schwefeldioxid, und manchmal auch Hydrogensulfid, sind am Geruch zu erkennen.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Geben Sie eine Zinkgranalie in ein trockenes Reagenzglas (keine Späne oder gar Pulver verwenden – beides reagiert zu heftig!). Geben Sie ca. 3 mL konzentrierte Schwefelsäure hinzu und erwärmen Sie die Mischung, bis eine merkliche Umsetzung unter Gasentwicklung einsetzt. Im oberen Teil des Reagenzglases bildet sich ein gelber Beschlag und gelbe Tröpfchen scheiden sich ab.
Formulieren Sie die Reaktionsgleichungen, jeweils eine für jedes entstehende Produkt. Welche Gefahr geht von dieser Reaktion aus?
Warum ist es unkorrekt alle beteiligten Reaktionspartner Schwefel, H2S und SO2 in einer Reaktionsgleichung zusammenzufassen (z.B.: 8 Zn + 3 H2SO4 + 16 H+ → 8 Zn2+ + S + H2S + SO2 + 10 H2O)?
Verdünnte und konzentrierte Schwefelsäure reagieren völlig verschieden. Woran mag das liegen?
Salpetersäure, HNO3
Konzentrierte Salpetersäure ist ähnlich aggressiv wie konzentrierte Schwefelsäure. In sehr verdünnter Form ist sie dagegen ein natürlich vorkommender Stoff: in Blitzen reagieren die Luftbestandteile Stickstoff und Sauerstoff zu „Stickoxiden“. Zusammen mit Regenwasser bildet sich dann Salpetersäure.
Eigenschaften
Salpetersäure ist in mehreren Konzentrationen laborüblich. Rauchende Salpetersäure ist ca. 95%ig. Sie ist durch Stickstoffdioxid gelb bis rotbraun gefärbt und gibt an der Luft Stickstoffdioxid-Dämpfe ab (daher auch „rote rauchende Salpetersäure“ genannt). Konzentrierte Salpetersäure ist ca. 69%ig, verdünnte Salpetersäure etwa 12%ig. Salpetersäure ist ein kräftiges Oxidationsmittel. Besonders die konzentrierten Lösungen sind sehr aggressiv. Ähnlich wie bei Schwefelsäure hängt auch das Verhalten von Salpetersäure gegenüber Metallen erheblich von der Säurekonzentration ab.
Handhabung
Der Umgang mit Salpetersäure (ab 5 %) wird durch eine Betriebsanweisung geregelt.
Reaktion von Zink mit konzentrierter Salpetersäure
Achten Sie beim Fortschreiten dieser Reaktion besonders auf deren schnell zunehmende Geschwindigkeit – ein wesentlicher Aspekt, wenn entgegen der Betriebsanweisung einmal viel größere Mengen der Reaktionspartner zusammenfinden.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
In ein Reagenzglas gebe man zu 1–2 mL konzentrierter Salpetersäure 1–2 Zinkgranalien (keine Späne oder gar Pulver verwenden – beides reagiert zu heftig!). Es tritt heftige Entwicklung von rotbraunem Stickstoffdioxid-Gas auf. Nachdem man dies beobachtet hat, wird die Reaktion durch Verdünnen mit viel Wasser beendet.
Stellen Sie die Reaktionsgleichung auf. Worin besteht die Gefahr? Warum wird die Reaktion immer heftiger? Was müsste man tun, um 1 kg Zink sicher zu Zinknitrat umzusetzen?
Wieviel Kilogramm Zinknitrat-Dihydrat lassen sich aus 1 kg Zink durch die Reaktion mit konzentrierter Salpetersäure herstellen?
Reaktion von Zink mit halbkonzentrierter Salpetersäure
Eine der Reaktionen, bei denen reichlich farbbloses NO gebildet wird, sodass Sie dessen spontane Oxidation zu braunem NO2 beim Kontakt mit Luft sehr schön beobachten können.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Man bereite in einem Reagenzglas durch Versetzen von etwas konzentrierter Salpetersäure mit etwas mehr als dem gleichen Volumen Wasser halbkonzentrierte Salpetersäure, gebe einige Zinkgranalien zu und erwärme. Anders als bei der Reaktion mit konzentrierter Säure entwickelt sich ein nur schwach braunes Gas: es entsteht ein Gemisch von viel farblosem Stickstoffmonoxid mit etwas braunem Stickstoffdioxid.
Stellen Sie die Reaktionsgleichung für die Bildung von NO auf. Worin besteht die Gefahr? Zur Wiederholung noch einmal: Warum wäre eine Reaktionsgleichung nicht korrekt, bei der auf der Seite der Produkte so etwas wie NO + NO2 stünde? Es entsteht doch wirklich nicht nur NO!
Viele Gase wie Wasserstoff, Methan oder Schwefeldioxid (aber auch feste und flüssige Substanzen), die mit Sauerstoff ein stabiles Reaktionsprodukt bilden, tun dies nicht bei Raumtemperatur. Es muss gezündet werden. Im Gegensatz dazu reagiert NO bei Raumtemperatur ungehemmt. Geben Sie eine Begründung dafür an.
Reaktion von Zink mit verdünnter Salpetersäure
Salzsäure, Schwefelsäure, Salpetersäure: jede dieser Säuren hat eine eigene Chemie – solange sie nicht verdünnt vorliegen. Vergleichen Sie selbst:
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
In einem Reagenzglas verdünne man etwas verdünnte Salpetersäure auf das doppelte Volumen, setzte einige Zinkgranalien zu und erwärme. Es entwickelt sich ein farbloses Gas, das sich auch bei Luftzutritt an der Mündung des Reagenzglases nicht braun färbt: mit der verdünnten Säure entsteht Wasserstoff.
Warum verlieren die Säuren beim Verdünnnen ihre Individualität?
Wieviel Kilogramm des als Beiz- und Bleichmittels verwendeten Zinknitrat-Hexahydrats lässt sich aus 1 kg Zink durch die Reaktion mit verdünnter Salpetersäure herstellen?
Reindarstellung von Stickstoffmonoxid
Umsetzungen, deren Gefahr in der unbeabsichtigten Freisetzung nitroser Gase besteht, lassen sich so steuern, dass eine besonders wichtige Verbindung gezielt und rein hergestellt werden kann: Stickstoffmonoxid, NO. Das großtechnische Zwischenprodukt bei der Herstellung von Salpetersäure aus Ammoniak ist zugleich ein Hormon, das zur Erweiterung der Blutgefäße führt.
Arbeiten Sie unbedingt unter einem gut ziehenden Abzug!
Geben Sie in den Rundkolben der für diesen Versuch zur Verfügung gestellten Gasentwicklungsapparatur eine Spatelspitze Kupferspäne und versetzen Sie diese mit einer Lösung aus zwei Teilen Wasser und einem Teil konzentrierter Salpetersäure. Nachdem die Luft im Reaktionsgefäß verdrängt ist, stülpe man über die Öffnung des Gasentbindungsrohres ein mit Wasser gefülltes Reagenzglas. Dabei beobachtet man im Kolben mehr oder weniger rotbraune Dämpfe, die ein Gemisch von Stickstoffmonoxid und Stickstoffdioxid darstellen. Beim Durchgang durch das Wasser reagiert Stickstoffdioxid. Das im Reagenzglas aufgefangene Gas besteht nur aus farblosem Stickstoffmonoxid. Hebt man das Reagenzglas aus dem Wasser heraus, so färbt sich der Inhalt von der Mündung her schnell braun, weil sich das Stickstoffmonoxid mit dem Luftsauerstoff zu Stickstoffdioxid umsetzt.
Formulieren Sie hier nicht nur die Reaktionsgleichung für die Umsetzung des Kupfers mit der Säure, sondern auch die Gleichung für die Reaktion des Stickstoffdioxids mit Wasser und für die Reaktion des Stickstoffmonoxids mit Luftsauerstoff.
Ammoniak, NH3
Die Grundchemikalie Ammoniak wird großtechnisch aus den Elementen über das Haber-Bosch-Verfahren gewonnen. In Form von Harnstoff und Ammoniumsalzen findet Ammoniak ausgedehnte Verwendung als Dünger. Als Ausgangsstoff für die Salpetersäureherstellung (Ostwald-Verfahren) erschließt Ammoniak den Einstieg in die technische Stickstoffchemie.
Eigenschaften
Während verdünnte Ammoniaklösung einen Gewichtsanteil von 10% aufweist, ist , konzentrierte Ammoniaklösung ist ca. 25%ig. Die konzentrierte Lösung hat eine Dichte von 0,91 g cm−3. Eine aufgrund der hohen Entweichungstendenz von Ammoniakgas kaum handhabbare gesättigte Lösung in Wasser ist mehr als 30%ig (Raumtemperatur).
Wieviel molar ist 25%ige Ammoniaklösung?
Handhabung
Der Umgang mit Ammoniaklösung wird durch eine Betriebsanweisung geregelt. Die dort als gefährlich aufgeführten Umsetzungen sind zum Teil so brisant, dass Sie diese – wie zum Beispiel die Umsetzung mit Iod zu Triiodnitrid („Iodstickstoff“, NI3) – nur in der Grundvorlesung kennenlernen werden, nicht aber in Praktikumsversuchen.
Ammoniak als Elektronenpaardonor
Einige der wichtigsten Konzepte der Chemie sind dem Ausgleich eines unterschiedlichen Angebots an negativer Ladungsdichte gewidmet. Ammoniak ist ein Lehrbuchbeispiel für ein Molekül, dessen Reaktionen durch die Anwesenheit eines freien Elektronenpaars am Stickstoffatom bestimmt sind. Stoffe, deren Reaktivität auf verfügbaren Elektronenpaaren beruht, sind in Säure-Base-Konzepten die Base, in Elektrophilie-Nukleophilie-Konzepten das Nukleophil und schließlich, als Spezialfall einer Säure-Base-Betrachtung, können sie gegenüber Metall-Ionen als Liganden auftreten.
Versetzen Sie etwa 2 mL destilliertes Wasser mit 2 Tropfen des Indikators Phenolphthalein. Geben Sie nun 1–2 Tropfen 6 m Ammoniaklösung zu. Versetzen Sie die jetzt rote Lösung mit einigen mL einer konzentrierten NH4Cl-Lösung, bis die rote Farbe wieder verschwindet.
Erklären Sie Ihre Beobachtung.
Stellen Sie eine Kupfersulfatlösung her, indem Sie ca. 2 Spatelspitzen Kupfer(II)-sulfat in etwa 2 mL destilliertem Wasser lösen. Tropfen Sie nun langsam 6 m Ammoniaklösung zu. Es bildet sich zunächst ein Niederschlag, der sich bei weiterer Zugabe von Ammoniaklösung allmählich wieder auflöst und eine tiefblaue Lösung entsteht.
Deuten Sie Ihre Beobachtungen durch Reaktionsgleichungen. Woher kommt die hellblaue Farbe der reinen Kupfersulfatlösung?
Natronlauge und Kalilauge, NaOH und KOH
„Natronlauge“ ist kein systematischer Name, sondern eine althergebrachte Bezeichnung für eine wässrige Lösung von Natriumhydroxid, NaOH, in Wasser. Dasselbe gilt für Kalilauge. Vor allem die konzentrierten Laugen sind deutlich gefährlicher als etwa eine gleichkonzentrierte Salzsäure, da sie viel hartnäckiger an der Haut haften als eine HCl-Lösung und nur schwer durch Wasser abgespült werden. Entsprechend gefährlich ist das heute vor allem auf historischen Jahrmärkten wieder in Mode gekommene „Seifensieden“ in offenen Bottichen: Mit den seit langem verfügbaren Chemikalien Branntkalk (Calciumoxid) oder Löschkalk (Calciumhydroxid) wird „Sodalösung“ (Natriumcarbonatlösung) „kaustifiziert“ (von lat. causticus ätzend, beizend). Anschließend wird die entstandene Lauge mit Fett umgesetzt, wobei dieses in Glycerin und Seife übergeht.
Haben Sie eine Idee, welche Chemie hinter dem Kaustifizieren und dem Seifensieden steckt?
Beim Auflösen von 40 g der handelsüblichen NaOH-Plätzchen in Wasser zu 1 L wird eine 1-molare Lösung erhalten. Beim Auflösen von 56 g KOH-Plätzchen zu 1 L wird jedoch eine nur 0,85-molare Lösung erhalten. Woran liegt das?
Eigenschaften
Verdünnte Natronlauge ist 7–8%ig (2 m), konzentrierte Natronlauge enthält 40 % Natriumhydroxid.
Handhabung
Der Umgang mit Alkalilaugen wird durch eine Betriebsanweisung geregelt.
Reaktion von Aluminium mit Natronlauge
Die Betriebsanweisung zu Alkalilaugen führt deren Umsetzung mit Metallen als besondere Gefahr auf. Der Versuch soll aufzeigen wieso.
Geben Sie eine Spatelspitze Aluminium-Grieß in ein Reagenzglas und stellen Sie dieses (vorsichtshalber) in ein hohes Becherglas. Geben Sie nun ein paar Tropfen verdünnte Natronlauge hinzu. Die Reaktion benötigt einen kurzen Augenblick bis sie in Gang kommt, verläuft dann aber äußert heftig (Aufschäumen des Reaktionsgemisches). Fangen Sie das entstehende Gas mit einem zweiten Reagenzglas auf und führen Sie eine Knallgasprobe durch.
Um welches Gas handelt es sich? Formulieren Sie die Reaktionsgleichung. Warum ist die Umsetzung mit Aluminium eine Gefahr?
Reaktion von Zink mit Natronlauge
Die Betriebsanweisung zu Alkalilaugen führt deren Umsetzung mit Metallen als besondere Gefahr auf. Der Versuch soll aufzeigen wieso.
Geben Sie eine Zinkgranalie in ein Reagenzglas und versetzen mit ein paar Tropfen verdünnte Natronlauge. Es kommt zu einer Gasentwicklung. Fangen Sie das entstehende Gas mit einem zweiten Reagenzglas auf und führen Sie eine Knallgasprobe durch.
Um welches Gas handelt es sich? Formulieren Sie die Reaktionsgleichung. Warum ist die Umsetzung mit Zink eine Gefahr?
Reaktion von Ammoniumsalzen mit Natronlauge
Die Betriebsanweisung zu Alkalilaugen führt deren Umsetzung mit Ammoniumsalzen als besondere Gefahr auf. Der Versuch soll aufzeigen wieso.
Geben Sie etwa vier Spatelspitzen Ammoniumchlorid in ein Reagenzglas und lösen Sie das Salz in ca. 2 mL destilliertem Wasser. Versetzen die Lösung mit ein paar Tropfen Natronlauge (6 m). Halten Sie anschließend ein angefeuchtetes pH-Papier an die Öffnung des Reagenzglases (ohne dieses mit dem Papier zu berühren).
Welches Gas können Sie so nachweisen (Reaktionsgleichung)? Warum führt die Betriebsanweisung die Reaktion mit Ammoniumsalzen als Gefahr auf?
Noch einmal, nur zur Wiederholung: Warum müssen sie darauf achten, das pH-Papier nicht das Reagenzglas berühren zu lassen?
Reaktionen von Metallsalzen mit Laugen
Versetzt man wässrige Lösungen von Übergangsmetallsalzen mit Natron- oder Kalilauge, beobachtet man zumeist die Bildung eines Niederschlags, es bildet sich ein Metallhydroxid. Einige dieser Hydroxide lassen sich nicht nur durch Zugabe von Säure wieder in Lösung bringen, sondern auch durch einen Überschuss an Lauge. Stoffe (im engeren Sinn Hydroxide), die sowohl als Säure als auch als Base reagieren können, bezeichnet man als amphoter.
Lösen Sie etwa zwei Spatelspitzen Aluminiumchlorid in 1–2 mL destilliertem Wasser und tropfen Sie langsam 6 m Natronlauge hinzu. Es bildet sich zunächst ein Niederschlag, der sich durch langsame Zugabe von einem Überschuss Natronlauge wieder auflöst. Geben Sie zu dieser klaren Lösung nun tropfenweise und unter gutem Durchmischen langsam 6 m Salzsäure, bis sich wieder ein Niederschlag bildet. Bei Zugabe von weiterer Säure löst sich dieser wieder auf.
Deuten Sie Ihre Beobachtungen durch Reaktionsgleichungen.
Wasserstoffperoxid-Lösung, H2O2
Eigenschaften
Wasserstoffperoxid-Lösung ist eine farblose Flüssigkeit, in der die gegen Zerfall in Wasser und Sauerstoff metastabile, blassblaue Flüssigkeit Wasserstoffperoxid in verdünnter Form vorliegt. Wasserstoffperoxid ist eine schwache Säure und gegenüber den meisten Stoffen ein starkes Oxidationsmittel, worauf die Verwendung als Bleich- und Desinfektionsmittel beruht. In hochkonzentrierter Form ist es sowohl als Einzel- als auch als Komponentenraketentreibstoff einsetzbar. Lösung in Wasser sind in mehreren Konzentrationen handelsüblich. Im Labor wird meist eine 30%ige Lösung verwendet, während im Alltag ca. 10%ige Lösung (zum Blondieren von Haaren) oder ca. 3%ige Lösung („Wasserstoffsuperoxid“, zur Desinfektion) eingesetzt wird.
Warum wird Wasserstoffperoxid, zum Beispiel bei der Papierherstellung (Zellstoffbleiche), als ein ökologisch günstig zu bewertendes („grünes“) Bleichmittel eingestuft?
In der Ausgabe vom 23. August 2008 berichtet die Süddeutsche im Münchner Teil:
Ätzende Chemikalie löst Feueralarm aus: In einem Versicherungsgebäude an der Dieselstraße ist am Donnerstagmittag das ätzende Gas Wasserstoffperoxid ausgetreten. Im zweiten Untergeschoss des Hauses entwich das Gas aus einem 30-Liter-Kunststoffbehälter, der die Chemikalie Bamacid enthielt. Diese wird zur Säuberung von Brauch- und Abwässern verwendet. [Anm.: Bamacid ist ein Entkeimungsmittel, das Wasserstoffperoxid und ein Wasserstoffperoxid-Derivat enthält.] Nachdem das Behältnis morgens gewechselt worden war, kam es aufgrund von Verunreinigungen zu einem Temperaturanstieg. Dadurch entwickelte sich ein Überdruck, der den Deckel absprengte. …
Können Sie den Hergang erklären? Wasserstoffperoxid ein Gas? Temperaturanstieg durch Verunreinigungen?
Handhabung
Der Umgang mit Wasserstoffperoxid-Lösung wird durch eine Betriebsanweisung geregelt.
Darstellung von Wasserstoffperoxid
Starke Säuren wie Schwefelsäure können das Anion von Peroxiden protonieren, wodurch Wasserstoffperoxid erhalten wird.
Führen Sie diesen Versuch paarweise aus.
Geben Sie in ein Becherglas oder Erlenmeyerkolben etwa 10 mL eiskalte 20%ige Schwefelsäure und fügen Sie in kleinen Mengen ca. 2 g Bariumperoxid hinzu. Um die Reaktion gut zu kühlen, geben sie während der Zugabe des Peroxids immer wieder kleine Eisstückchen dazu. Während der Reaktion fällt schwerlösliches Bariumsulfat aus. Nach der vollständigen Zugabes des Bariumperoxides wird mit festem Bariumcarbonat versetzt, damit die überschüssige Säure abreagiert. Filtrieren Sie anschließend das Reaktionsgemisch und benutzen Sie die so erhaltene Wasserstoffperoxid-Lösung in den folgenden Versuchen.
Formulieren Sie die Reaktionsgleichungen.
Oxidierende Eigenschaft von Wasserstoffperoxid
Bringen Sie ca. 2 mL der im vorherigen Versuch hergestellten Wasserstoffperoxid-Lösung mit Natronlauge auf einen pH-Wert des stark basischen Bereiches und fügen Sie einige Tropfen einer MnSO4-Lösung zu.
Was können Sie beobachten?
Sauerstoff aus Wasserstoffperoxid
Wasserstoffperoxid kann als Quelle zur Synthese kleiner Mengen reinen Sauerstoffs genutzt werden.
Geben Sie eine Spatelspitze Braunstein, MnO2, in ein Reagenzglas und tropfen Sie vorsichtig ca. 0,5 mL der zuvor hergestellten Wasserstoffperoxid-Lösung hinzu. Es kommt zu einer Gasentwicklung. Führen Sie eine Glimmspannprobe durch, um einen Hinweis auf die Identität des Gases zu bekommen.
Welches Gas haben Sie nachgewiesen? Welche Funktion hat MnO2 bei dieser Reaktion? Formulieren Sie die Reaktionsgleichung.
Maßanalyse
Im Mittelpunkt des Grundkurses stehen zwei handwerkliche Aspekte: Sicheres Arbeiten und genaues Arbeiten. Letzteres lernen Sie im Kapitel „Maßanalyse“ kennen. Ziel ist das genaue Abmessen von Volumina und genaues Wiegen.
Messgefäße
Die Maßanalyse beruht auf der präzisen Bestimmung von Flüssigkeitsvolumina. Die hierbei verwendeten Volumenmessgeräte sind Messzylinder, Messkolben, Pipetten und Büretten. Der Inhalt der Messgefäße wird in mL angegeben und ist auf eine Temperatur von 20 °C geeicht. Messkolben und Messzylinder sind auf Einguss (Kennzeichnung „In“) geeicht, das heißt, dass sie bei der entsprechenden Eichtemperatur das angegebene Volumen fassen. Büretten und Pipetten sind dagegen in der Regel auf Auslauf (Kennzeichnung „Ex“) geeicht, das heißt, dass beim Entleeren der auf dem Messgefäß angegebene Inhalt abgegeben wird. Geeichte Messgefäße werden (nach der Deutschen Eichordnung) in Geräte der Klassen A und B unterteilt, wobei die Geräte der Klasse B eine doppelt so große Fehlergrenze besitzen. Um welche Klasse es sich handelt, erkennen Sie an der Kennzeichnung „A“ bzw. „B“ auf dem jeweiligen Messgefäß.
Messkolben
Messkolben sind Standkolben mit einem langen, engen Hals, auf dem sich eine Eichmarke befindet. Sie dienen vor allem zur Herstellung von Maßlösungen. Befüllen Sie dazu den Messkolben zunächst mit einer genau abgewogenen Substanzmenge oder mit einem Standardkonzentrat und füllen Sie den Kolben mit destilliertem Wasser bis zur Hälfte auf. Lösen oder durchmischen Sie nun die Substanz durch vorsichtiges Schwenken. Um das Volumen so exakt wie möglich abzumessen, befüllen Sie den Kolben nun zunächst bis etwa ein oder zwei Zentimeter unterhalb der Eichmarke. Warten Sie dann 1–2 Minuten, damit gegebenenfalls Flüssigkeitsreste von der Glaswand ablaufen können. Verwenden Sie dann eine Einwegpipette und geben Sie gerade so viel Flüssigkeit hinzu, bis der unterste Punkt des Flüssigkeitsspiegels (der „Meniskus“) mit der Eichmarke zusammenfällt. Die Markierung muss beim Ablesen auf Augenhöhe liegen, so dass der vordere Teil des Ringes den hinteren genau überdeckt. Verschließen Sie anschließend den Messkolben und schütteln Sie ihn über Kopf, um die Lösung gut zu durchmischen.
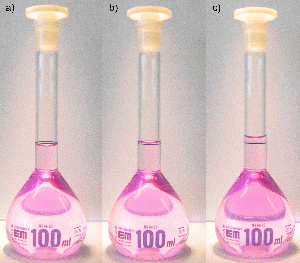
Befüllen eines Messkolbens: a) zu wenig, b) richtig befüllt, c) viel zu viel.
Messzylinder
Messzylinder sind Standzylinder, auf denen sich eine Graduierung befindet, mit deren Hilfe sich ein bestimmtes Volumen abmessen lässt. Messzylinder sind weniger genau als Messkolben und sollten daher nur zur Grobmessung verwendet werden. (Unter keinen Umständen sollten Sie Bechergläser oder Erlenmeyerkolben zur Abmessung von Flüssigkeitsmengen zur Analyse verwenden, da die Volumenmarkierung bei diesen Geräten nur als grober Anhaltspunkt angebracht sind.) Achten Sie beim Abmessen auch hier darauf, dass die Markierung für das gewünschte Volumen mit dem untersten Punkt des Flüssigkeitsspiegels zusammenfällt und dass Sie diese Markierung in Augenhöhe betrachten.
Pipetten
Pipetten lassen sich in Voll- und Messpipetten unterteilen. Vollpipetten sind Glasröhren mit einer Ausbuchtung in der Mitte. Ihr Inhalt wird durch eine Eichmarke am oberen Ende markiert. Messpipetten sind Glasröhren, die im Unterschied zu den Vollpipetten eine Graduierung besitzen.
Zur Abmessung eines gewünschten Volumens tauchen Sie die Spitze der Pipette in die entsprechende Flüssigkeit und ziehen diese bis über die Eichmarke auf. Legen Sie die Pipettenspitze an der Glaswand des schräg gehaltenen Gefäßes an und lassen Sie so viel Flüssigkeit herauslaufen, bis der Meniskus auf der Eichmarke liegt. Wichtig ist, dass Sie die Pipette dabei gerade halten und die Eichmarke auf Augenhöhe liegt. Streifen Sie nun die Pipettenspitze an der Glaswand ab und entleeren Sie die Flüssigkeit in das vorgesehene Gefäß. Dabei liegt die Pipettenspitze wieder an der Glaswand des schräg gehaltenen Gefäßes an. Nachdem die Pipette abgelaufen ist, warten Sie noch solange wie für den Pipettentyp vorgesehen (siehe unten), bevor Sie die Pipette nach oben hin von der Gefäßwand abziehen. Da Pipetten auf Auslauf geeicht sind, wurde nun der auf der Pipette angegebenen Inhalt abgegeben. Dass in der Pipettenspitze ein Rest zurückbleibt, ist bei der Eichung auf Auslauf berücksichtigt, so dass dieser nicht, zum Beispiel durch Ausblasen, zusätzlich entleert werden darf.
Als Pipettierhilfe zum Aufsaugen und Ablassen der Flüssigkeit wird ein Peleusball verwendet. Dieser besitzt drei Ventile. Ventil A dient zum Ausdrücken der Luft aus dem Ball, Ventil S zum Ansaugen der Flüssigkeit und Ventil E zum Entleeren der Pipette. Der Peleusball wird auf die Pipette gesetzt, Ventil A durch Zusammendrücken mit Daumen und Zeigefinger geöffnet und die Luft aus dem Gummiball gedrückt. Durch Druck auf Ventil S kann nun die Flüssigkeit aufgezogen und durch Druck auf Ventil E wieder abgelassen werden.

Peleusball
Mess- und Vollpipetten werden in unterschiedliche Genauigkeitsklassen eingeteilt. Bei Pipetten der Klasse A ist die Ablaufzeit durch eine verengte Spitze verlängert, sodass bereits während des Ablaufens die Flüssigkeit von der Glaswand nachläuft. Die Pipetten der Klasse B besitzen eine erheblich kürzere Ablaufzeit, dafür aber auch eine doppelt so große Fehlergrenze. Neben den Typen A und B gibt es noch Pipetten der Klasse AS. Es handelt sich dabei um Pipetten mit der gleichen Fehlergrenze wie jene der Klasse A, aber mit deutlich kürzeren Ablaufzeiten. Jedoch benötigen diese nach dem Ablauf der Flüssigkeit eine kurze Wartezeit, meist 15 Sekunden. Die genaue Wartezeit finden Sie auf der jeweiligen Pipette des Typs AS, z.B. „Ex+15s“ für eine Wartezeit von 15 Sekunden nach dem Ablauf der Flüssigkeit. Pipetten der Klassen A und B benötigen keine Wartezeit. Zur Übersicht finden Sie die Fehlergrenzen und Ablaufzeiten ausgewählter Vollpipetten in der folgenden Tabelle.
| Klasse A | Klasse AS | Klasse B | ||||
|---|---|---|---|---|---|---|
| Nennvolumen [mL] | max. Fehler [%] | Ablaufzeit [s] | max. Fehler [%] | Ablaufzeit [s] | max. Fehler [%] | Ablaufzeit [s] |
| 5 | ± 0,30 | 15–30 | ± 0,30 | 7–11 | ± 0,60 | 7–30 |
| 10 | ± 0,20 | 15–40 | ± 0,20 | 8–12 | ± 0,40 | 8–40 |
| 20 | ± 0,15 | 25–50 | ± 0,15 | 9–13 | ± 0,30 | 9–50 |
| 25 | ± 0,13 | 25–50 | ± 0,13 | 10–15 | ± 0,26 | 10–50 |
| 50 | ± 0,10 | 30–60 | ± 0,10 | 13–18 | ± 0,20 | 13–60 |
| 100 | ± 0,08 | 40–60 | ± 0,08 | 25–30 | ± 0,16 | 25–60 |
Beachten Sie, dass im Praktikum möglichst genaues Arbeiten geübt werden soll. Verwenden Sie daher Pipetten der Typen A oder AS und beachten Sie die angegebene Wartezeit.
Büretten
Büretten sind graduierte Glasrohre mit einem Hahn am unteren Ende zur regelbaren Abgabe genau bekannter Flüssigkeitsmengen. Zur Benutzung der Bürette spannen Sie diese senkrecht in ein Stativ ein und befüllen sie mit Hilfe eines Trichters zunächst mit ein paar Millilitern der Flüssigkeit. Öffnen sie vorsichtig den Hahn bis die Flüssigkeit herauszulaufen beginnt und schließen Sie den Hahn dann wieder. Die Ablaufspitze sollte jetzt vollständig mit Flüssigkeit gefüllt sein, es darf sich unter keinen Umständen noch Luft im unteren Teil der Bürette befinden. Füllen Sie die Bürette nun mit Flüssigkeit bis ca. 0,5 cm oberhalb der Nullmarke auf und lassen Sie durch vorsichtiges Öffnen des Hahns die Flüssigkeit bis zur Nullmarke ab. Wie bei den anderen Messgefäßen wird auch hier der Flüssigkeitsstand an der tiefsten Stelle des Meniskus abgelesen. Das Ablesen wird dabei durch einen farbigen Streifen (Schellbachstreifen) auf der Rückwand der Bürette erleichtert. Auf der Höhe des Flüssigkeitsspiegels ist dieser Streifen fast vollständig eingeschnürt. Eine Ausnahme bilden intensiv farbige Flüssigkeiten, bei denen an der oberen Kante ablesen wird.
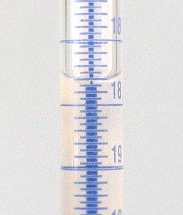
Schellbachstreifen
Sind Sie am Endpunkt einer Titration angelangt, wird sich der Meniskus in den seltensten Fällen genau auf einem Teilstrich der Graduierung befinden. Orientieren Sie sich in einem solchen Fall an den Markierungen ober- und unterhalb des Meniskus und schätzen Sie den Wert dazwischen so genau wie eben möglich auf die zweite Nachkommastelle ab. Wenn Sie z.B. eine 25-mL-Bürette mit Teilstrichen von 0,05 mL verwenden und der Meniskus zwischen 11,15 und 11,20 mL liegt, so schätzen Sie ab, ob er bei 11,16, 11,17, 11,18 oder 11,19 mL befindet.
Wird eine Bürette mit Glashahn benutzt, so muss dieser vor der Benutzung leicht gefettet werden (dies entfällt bei einem Teflonhahn). Achten Sie darauf, dass kein Schlifffett in die Hahnbohrung gelangt. Der Schliff sollte gleichmäßig klar erscheinen und der Hahn leicht drehbar sein. Verwenden Sie zum Fetten nur das dafür geeignete Schlifffett.
Nehmen Sie einen 250-mL-Messkolben zur Hand und entleeren Sie in diesen fünfmal eine 50 mL Vollpipette mit destilliertem Wasser.
Konnten Sie den Messkolben exakt befüllen? Falls nicht, überlegen Sie, wieso Sie die Eichmarke nicht genau getroffen haben.
Befüllen Sie einen 250-mL-Messkolben mit gekühltem Wasser und beobachten Sie was passiert, wenn das Wasser Raumtemperatur erreicht hat.
Erklären Sie Ihre Beobachtung.
Reinigung der Messgefäße
Wie wichtig eine gründliche Reinigung von Laborgeräten ist, haben Sie bereits in einem früheren Versuch gelernt. Dies gilt besonders für die zur quantitativen Analyse verwendeten Messgefäße, die sowohl frei von chemischen Rückständen, als auch frei von Staub und Fett sein müssen. Ein Fettfilm in Büretten oder Pipetten führt zu einer geringeren Benetzung der Glaswand und infolge dessen zu einer Vergrößerung des abgegebenen Volumens.
Reinigen Sie daher ihre Glasgeräte immer unmittelbar nach dem Gebrauch, indem Sie gründlich mit destilliertem Wasser spülen. Verwenden Sie verdünnte Säuren oder Laugen nur bei gröberen Verschmutzungen, die sich sonst nicht beseitigen lassen. Konzentrierte Laugen sollten nicht verwendet werden, da diese über einen längeren Zeitraum das Glas angreifen. Spülen Sie ihre Messgefäße keinesfalls mit Aceton, Ethanol oder einem anderen organischen Lösemittel nach, da diese Fettspuren enthalten können, welche dann auf der Glaswand zurückbleiben. Auch das Verwenden von Druckluft zum schnelleren Trocknen der Messgefäße ist zu unterlassen, da hierbei Fett aus den Leitungen in die Glasgeräte gelangen kann. Vor der Verwendung Ihrer Bürette sollten Sie diese mit einigen Millilitern der Titrationslösung spülen, um eventuelle Rückstände oder Staub zu entfernen.
Indikatoren
Zur Maßanalyse zählt die Detektion des Endpunktes einer chemischen Reaktion – hier die Detektion der vollständigen Reaktion einer Säure mit einer Base. Sie lernen in der begleitenden Vorlesung, dass die hierzu geeigneten Indikatoren (lat. indicare anzeigen) selbst Säuren und Basen sind und so Änderungen des pH-Wertes auf der Grundlage des folgenden Protolysegleichgewichts anzeigen können:
HInd + H2O ⇌ H3O+ + Ind−
Zu etwa 10 mL Wasser werden einige Tropfen Methylorange-Lösung gegeben. Die Lösung wird tropfenweise mit verdünnter Salzsäure versetzt, wobei sich die Farbe in charakteristischer Weise ändert. Nun wird bis zum Farbumschlag tropfenweise verdünnte Natronlauge zugegeben.
Zu etwa 10 mL Wasser werden einige Tropfen Phenolphthalein-Lösung gegeben. Die Lösung wird tropfenweise mit verdünnter Natronlauge versetzt, wobei sich die Farbe in charakteristischer Weise ändert. Nun wird bis zum Farbumschlag tropfenweise verdünnte Salzsäure zugegeben. Versuchen Sie eine Mischfarbe zu erreichen.
Maßlösungen
Lösungen mit genau bekanntem Gehalt (Titer) an Gelöstem heißen Maßlösungen oder eingestellte Lösungen. Die genaue Konzentration wird durch die Angabe der ungefähren Konzentration (zum Beispiel 0,1 m) und der Angabe eines in der Regel geringfügig von 1 abweichenden Faktors angegeben.
Beispiel: Eine Natronlauge der Konzentration 0,0996 m ist eine 0,1 m Maßlösung mit dem Faktor 0,996. Sie wird eingesetzt, wenn eine Vorschrift eine 0,1 m Natronlauge verlangt.
Herstellung einer annähernd 0,1 m Natronlauge-Lösung
Zur Herstellung einer 0,1 m Natronlauge wiegen Sie eine genaue Menge an Natriumhydroxid (wie viel?) ab. Spülen Sie die NaOH-Plätzchen mit etwas destilliertem Wasser ab um die an der Oberfläche anhaftende Carbonatschicht zu entfernen. Lösen Sie dann das Natriumhydroxid in einem ausreichend großem Becherglas und füllen Sie die Lösung mit Hilfe eines Trichters in einen 1-L-Messkolben. Spülen Sie Reste der NaOH-Lösung im Becherglas mit etwas destilliertem Wasser in den Messkolben und füllen Sie diesen dann bis zur Eichmarkierung auf. Lagern sie Ihre Natronlauge stets gut verschlossen in einer Polyethylenflasche, damit diese möglichst kein CO2 aus der Luft aufnimmt.
Durchführung einer Titration
Die zu bestimmende Lösung erhalten Sie in einem Messkolben (250 mL), den Sie mit destilliertem Wasser wie oben beschrieben bis zur Eichmarke auffüllen. Entnehmen Sie der Lösung nun eine genau definierte Menge mit Hilfe einer Vollpipette (in der Regel 25 oder 50 mL) und entleeren Sie diese in einen Weithals-Erlenmeyerkolben mit ausreichendem Volumen. Verdünnen Sie ihre Probelösung mit destilliertem Wasser auf etwa das doppelte Volumen und setzen Sie anschließend den für die jeweilige Titration vorgesehenen Indikator zu. Dabei reichen im Normalfall einige Tropfen der Indikatorlösung. Beachten Sie gegebenenfalls weitere Titrationsbedingungen wie zum Beispiel den pH-Wert.
Füllen Sie nun Ihre Bürette mit der entsprechenden Titratorflüssigkeit (Maßlösung) und notieren Sie den Flüssigkeitsstand (Nullmarke; muss nicht zwingend auf der 0-mL-Marke der Eichskala liegen, natürlich keinesfalls darüber). Nun können Sie mit dem Eintropfen der Maßlösung in Ihre Probelösung beginnen, wobei Sie durch andauerndes Schwenken des Erlenmeyerkolbens für eine gute Durchmischung sorgen. Zu Beginn der Titration können Sie die Maßlösung noch etwas schneller zutropfen, gegen Ende jedoch nur noch tropfenweise. Achten Sie auf einen an der Bürettenspitze hängenden Tropfen. Ein solcher Tropfen gehört zu Ihrer verbrauchten Maßlösung. Sie können Ihn an der Glaswand des Erlenmeyerkolbens abstreifen und mit Hilfe von wenig(!) destilliertem Wasser in die Probelösung spülen.
Warten Sie nach erfolgtem Farbumschlag die Ablaufzeit der Bürette ab und lesen Sie dann den Flüssigkeitsstand ab. Es kann mitunter hilfreich sein, den Flüssigkeitsstand schon kurz vor dem vermuteten Endpunkt zu notieren, um im Falle eines Übertitrierens noch ein brauchbares Ergebnis zu erhalten. Eine weiße Unterlage, zum Beispiel ein Blatt Papier, kann die Erkennung des Umschlagspunktes erleichtern. Auch eine Vergleichslösung kann helfen, den Umschlagspunkt besser zu erkennen. Machen sie sich zudem immer schon vorher bewusst, bei welchem Verbrauch an Maßlösung Sie mit dem Endpunkt der Titration rechnen dürfen, um rechtzeitig vom stetigen zum langsamen Eintropfen überzugehen.
Wichtig: Führen Sie, um ein möglichst exaktes Ergebnis zu erhalten, immer mehrere Titrationen unter den gleichen Bedingungen durch und bilden Sie den Mittelwert aus den einzelnen Messwerten. Weicht ein Ergebnis deutlich von den anderen ab, sollten Sie dieses nicht in Ihre Berechnung mit einbeziehen.
Faktorbestimmung
Die erste Titration in diesem Grundkurs dient zur Faktorbestimmung der Natronlauge aus dem vorhergehenden Versuch. Um den Faktor der Natronlauge zu bestimmen, gibt es zwei Möglichkeiten. Man kann die Natronlauge mit einer bereits eingestellten Säure titrieren oder man setzt die Natronlauge mit der Lösung eines geeigneten Urtiters um. Zum Einstellen von Natronlauge benutzt man im Allgemeinen Kaliumhydrogenphthalat als Urtitersubstanz.
Stellen Sie, um den Faktor der Natronlauge aus dem vorhergehenden zu bestimmen, eine ca. 0,05 m Kaliumhydrogenphthalat-Lösung her. Trocknen Sie dazu das Kaliumhydrogenphthalat im Trockenschrank vor (ca. 30 min bei 110 °C) und wiegen Sie die benötigte Menge möglichst genau ein und notieren Sie den genauen Wert. Überführen Sie die eingewogene Menge in einen 250-mL-Messkolben und befüllen Sie diesen zunächst etwa zur Hälfte mit destilliertem Wasser. Lösen Sie das Kaliumhydrogenphthalat durch Schütteln des Messkolbens und füllen Sie dann bis zur Eichmarke auf. Füllen Sie nun mit einer Vollpipette 25 mL der Urtiterlösung in einen Erlenmeyerkolben, verdünnen Sie mit destilliertem Wasser auf ca. 100 mL und setzen Sie einige Tropfen Phenolphthalein als Indikator zu. Füllen Sie Ihre Bürette mit der einzustellenden Natronlauge und titrieren Sie bis zum Farbumschlag nach leicht rosa.
Führen Sie die Titration mindestens dreimal durch und errechnen Sie aus dem Verbrauch der NaOH-Lösung deren Konzentration.
Alkalimetrie
Alkalimetrische Titration einer einprotonigen Säure: Salzsäure
Pipettieren Sie mit einer Vollpipette 25 mL Ihrer Probenlösung in einen Erlenmeyerkolben, verdünnen Sie auf ca. 100 mL und durchmischen Sie das Ganze durch Schwenken des Kolbens. Geben Sie ein paar Tropfen Methylorange hinzu und titrieren Sie mit der im vorherigen Versuch eingestellten Natronlauge bis zum Farbumschlag von rot nach gelb. Notieren Sie den Verbrauch an Maßlösung und berechnen Sie die Gesamtmenge an HCl in der Probenlösung. Führen Sie die Titration mindestens dreimal durch.
Weshalb sind für die Titration von Salzsäure mit Natronlauge alle Indikatoren mit Umschlagspunkten zwischen pH 4 und pH 10 geeignet, für die Titration von Essigsäure mit Natronlauge jedoch nicht?
Farbe
Warum ist ein grünes Leuchten in zahlreichen Fernsehkriminalfällen ein Indiz für ein Verbrechen? Warum brennen Lithium-Salze mit roter Flamme und warum erscheint eine Suspension von Goldnanopartikeln blau? Woher kommen diese Farbeindrücke? Farbreaktionen und -eindrücke bestimmen das tägliche Leben und sind auch ein wesentlicher Bestandteil der Analytischen Chemie. Tatsächlich haben „Farben“ ganz verschiedenen Ursachen. In diesem Projekt werden grundlegende Phänomene, die zu Farbeindrücken führen, unterschieden und Messmethoden vorgestellt. Die erarbeiteten physiko-chemischen Kenntnisse bilden die Grundlage für sämtliche weiteren Praktika in den verschiedenen Lehrbereichen.
Wissenswertes vorab
Licht: Besteht aus elektromagnetischen Wellen, die Energie mit bestimmter Wellenlänge und Ausbreitungsrichtung transportieren.
Absorption: Absorption von sichtbarem Licht führt zu einer elektronischen Anregung der Materie, die Intensität des einfallenden Lichts wird dadurch abgeschwächt.
Fluoreszenz: Nach Absorption von Licht können manche Materialien die aufgenommene Energie in Form von sichtbarem Licht, als Fluoreszenz oder Phosphoreszenz, wieder abgeben.
Streuung: Elastische Lichtstreuung führt nur zu einer Änderung der Ausbreitungsrichtung.
Optische Spektroskopie: Absorption, Fluoreszenz und Streuung einer Probe sind stoffspezifische Eigenschaften. Die spektral aufgelöste Bestimmung dieser Eigenschaften durch die optische Spektroskopie ermöglicht somit die zerstörungsfreie Analyse von Stoffen.
Farbreaktionen: Chemische Reaktionen, bei deren Verlauf eine Farbänderung auftritt.
Lernziele und einführende Literatur
Lernziele: Ursache von Farbeindrücken, Licht als elektromagnetische Welle und Licht als Photonen, Lichtintensität als Funktion der Wellenlänge, Spektrum, Absorption, Streuung, Fluoreszenz, Extinktion, Spektrometer, UV/Vis-Spektroskopie, Lambert-Beer-Gesetz, Flammenfärbung, dispersive Elemente, Polarisator, optische Filter, additive/subtraktive Farbmischung, Polarisation, Kontinuum, Linienspektrum.
Einführende Literatur: Vorlesungsskript, Atkins
Vorversuche
Spektrale Zerlegung einer Weiß-Lichtquelle
Weißes Licht besteht aus verschiedenen Spektralanteilen, die mittels eines dispersiven Elements räumlich aufgespalten werden können. Dispersive Elemente sind beispielsweise Prismen oder optische Beugungsgitter. Auch feine Wassertropfen können Licht in seine spektralen Bestandteile aufteilen und so einen Regenbogen verursachen.
Im ersten Vorversuch sollen die Funktionsweisen verschiedener dispersiver Elemente untersucht werden.
Untersuchen Sie die optischen Eigenschaften eines Prismas, eines Beugungsgitters und eines Handspektroskops mit Hilfe verschiedener Lichtquellen (z. B. Neonröhre, Sonne, Taschenlampe). Betrachten Sie dazu das ausfallende bzw. reflektierte Licht unter verschiedenen Winkeln. Variieren Sie auch den Einfallswinkel. Betrachten sie des Weiteren die Reflexion einer Taschenlampe auf der Unterseite einer CD und einer DVD.
Skizzieren sie den Strahlengang des Lichts durch das Prisma und die Aufspaltung des Lichts. [2P]
Aus welchen sichtbaren Spektralanteilen besteht das Licht der verschiedenen Lichtquellen? [2P]
Was lässt sich bei der Beleuchtung einer CD-Unterseite/DVD-Unterseite mit der Taschenlampe beobachten? [1P]
Welche Unterschiede zwischen der CD und der DVD lassen sich erkennen und was ist die Ursache hierfür? [2P]
Spektrale Analyse verschiedener Lichtquellen
Der Vorversuch dient dem Verständnis der Eigenschaften und Unterschiede zwischen einem Linienspektrum und einem Kontinuum.
Führen Sie eine spektrale Analyse des Lichts einer Neonröhre, einer Taschenlampe und einer LED durch. Verwenden Sie hierzu ein Faserspektrometer und ein Handspektroskop.
Verwenden Sie dazu das Spektrometer im Scope-Modus
![]() mit einer Integrationszeit von 3,8 ms, 10 Scans zur Mittelwertbildung und einer Boxcar Breite von 2.
mit einer Integrationszeit von 3,8 ms, 10 Scans zur Mittelwertbildung und einer Boxcar Breite von 2.
Achten Sie darauf, dass die Ausleuchtung des Spektrometers 65000 Counts nicht übersteigt! Richten Sie deshalb das offene Ende der Faser nie direkt in die Lichtquelle, sondern nähern diese vorsichtig von der Seite an.
Stellen Sie die Spektren der verschiedenen Lichtquellen in Graphen dar. [2P]
Wodurch unterscheiden sich die Spektren der Lichtquellen?
Wann erscheint eine Lichtquelle „weiß“, wann mit einer „bestimmten Farbe“? [2P]
Polarisation
Das Licht einer natürlichen Quelle ist unpolarisiert. Polarisatoren dienen zur Selektion bestimmter Polarisationsebenen und können somit auch als Analysatoren verwendet werden. Optisch aktive Substanzen wie beispielsweise D(+)-Glucose können die Polarisationsrichtung des Lichtes drehen. Diese Eigenschaft kann als Nachweis für die Reinheit von optisch aktiven Substanzen verwendet werden.
Betrachten Sie eine Lichtquelle durch zwei Folienpolarisatoren während Sie einen der beiden drehen.
Überprüfen Sie die Formel aus der Vorlesung für die Transmission als Funktion des Winkels zwischen den beiden Polarisatoren qualitativ. [2P]
Betrachten Sie das Licht einer Deckenlampe durch einen Folienpolarisator nachdem es an einer glatten Oberfläche reflektiert wurde. Versuchen Sie dabei eine Auslöschung des reflektierten Lichts zu beobachten.
Begründen Sie Ihre Beobachtungen im Fall des reflektierten Lichts. Was kann hieraus über die Lichtausbreitung gefolgert werden? [2P]
Wie kann aus diesem Versuch die Transmissionsrichtung des Analysators bestimmt werden? [1P]
Extinktion, Absorption und Transmission verschiedener Farbfilter und farbiger Lösungen
Es werden die Effekte von RGB und CMY Farbfiltern untersucht. Dabei wird der Zusammenhang zwischen Farbeindruck und Spektrum betrachtet. Die Wirkung von farbigen Lösungen als Farbfilter wird anhand eines pH-Indikators bei verschiedenen pH Werten betrachtet.
Die Begriffe Extinktion, Absorption und Transmission werden eingeführt und unterschieden.
Messen Sie qualitativ die Absorptionsspektren aller 6 Farbfilter mit Hilfe des UV/Vis-Spektrometers. Verwenden Sie dazu das Spektrometer im Scope-Modus ![]() mit einer Integrationszeit von 3,8 ms, 10 Scans zur Mittelwertbildung und einer Boxcar Breite von 2.
mit einer Integrationszeit von 3,8 ms, 10 Scans zur Mittelwertbildung und einer Boxcar Breite von 2.
Achten Sie darauf, dass die Ausleuchtung des Spektrometers 65000 Counts nicht übersteigt! Richten Sie deshalb das offene Ende der Faser nie direkt in die Lichtquelle, sondern nähern diese vorsichtig von der Seite an.
Verwenden Sie als Lichtquelle eine Taschenlampe und nehmen Sie davon zunächst ein Spektrum auf. Messen Sie nun für jeden Farbfilter jeweils ein Spektrum, indem Sie den Farbfilter zwischen Taschenlampe und dem offenen Ende der Faser einbringen.
Stellen Sie die Spektren der RGB Filter zusammen mit dem der Taschenlampe in einem Graphen dar. Erstellen Sie einen zweiten Graphen entsprechend mit den Spektren der CMY Filter und der Taschenlampe. [2P]
Wie entsteht der Farbeindruck nach einem Farbfilter? Erläutern Sie den Unterschied zwischen den RGB und den CMY Filtern jeweils anhand eines Beispiels. [2P]
Zur Aufnahme von Absorptionsspektren verschiedener Indikatorlösungen verfahren Sie wie im Abschnitt Aufnahme von Absorptionsspektren erläutert ist. Verwenden Sie jeweils eine basische und eine saure Lösung von Methylorange (je 3 mL einer 0,1 m HCl-Lösung und einer 0,1 m NaOH-Lösung
mit je einem Tropfen Indikator versetzt).
Stellen Sie die beiden Absorptionsspektren in einem Graphen dar. Wo liegen die Absorptionsmaxima für Methylorange im Basischen und im Sauren? [1P]
Berechnen Sie anhand der beiden Absorptionsspektren die Transmissionsspektren der Indikatorlösungen. [2P]
Stellen Sie die Reaktionsgleichung für die Reaktion in saurer und basischer Umgebung für Methylorange auf. [1P]
Fluoreszenz
Der Zusammenhang zwischen der Konzentration einer Fluoreszenzfarbstofflösung und der Intensität des Emissionsmaximums soll unter Verwendung eines Fluoreszenzspektrometers untersucht werden.
Betrachten Sie zunächst eine konzentrierte Lösung von Fluorescein aus dem Indikatorsatz.
Welchen Farbeindruck erhalten Sie bei der Betrachtung der konzentrierten Fluoresceinlösung? [1P]
Gegeben ist eine Ausgangslösung von Fluorescein in Ethanol bekannter Konzentration. Stellen Sie eine Verdünnungsreihe mit den Konzentrationen 0,1 mmol L−1, 0,2 mmol L−1, 0,3 mmol L−1, 0,4 mmol L−1, 0,6 mmol L−1, 0,8 mmol L−1 und 1,0 mmol L−1 her und messen Sie die Fluoreszenzemissionsspektren. Die Küvette sollte dabei für jede Verdünnungsstufe auf ein Gesamtvolumen von 2 mL aufgefüllt werden.
Stellen Sie die gemessenen Fluoreszenzspektren in einem Graphen dar. Entnehmen Sie aus den Spektren die Wellenlänge des Emissionsmaximums. Was wäre eine geeignete Anregungswellenlänge? [3P]
Entnehmen Sie an der Wellenläge des zuvor ermittelten Emissionsmaximums die Intensität bei verschiedenen Konzentrationen. Erstellen Sie daraus ein Diagramm, indem Sie die Intensität gegen die Konzentration auftragen. Bei welchen Konzentrationen befindet man sich bezüglich der Fluoreszenzintensität im linearen Bereich? Woraus resultieren Abweichungen vom linearen Verhalten? [3P]
Übungsanalysen
Anwendung des Lambert-Beerschen Gesetzes
Mit Hilfe des Lambert-Beerschen Gesetzes wird der Extinktionskoeffizient eines Farbstoffs aus der gemessenen Optischen Dichte bei verschiedenen Konzentrationen ermittelt.
Gegeben ist eine 1,0 mmol L−1 Stammlösung von Chinolingelb in Wasser.
Stellen Sie eine Verdünnungsreihe mit den Konzentrationen 0,005 mmol L−1, 0,01 mmol L−1, 0,02 mmol L−1, 0,03 mmol L−1, 0,04 mmol L−1 her. Die Küvette sollte dabei für jede Verdünnungsstufe auf ein Gesamtvolumen von 2 mL aufgefüllt werden. Messen Sie von allen Verdünnungen jeweils ein Absorptionsspektrum.
Stellen Sie die gemessenen Spektren in einem Graphen dar und bestimmen sie die Wellenlänge des Absorptionsmaximums. [2P]
Fertigen Sie ein Diagramm an, indem Sie die Optische Dichte an der Wellenlänge des zuvor bestimmten Absorptionsmaximums gegen die Konzentration auftragen. Passen Sie eine Ursprungsgerade an die Datenpunkte an. Ermitteln Sie den Extinktionskoeffizienten von Chinolingelb mit Hilfe des Lambert-Beerschen Gesetzes aus der Steigung der Regressionsgeraden (Einheit beachten!). [3P]
Warum wird für die Anpassung eine Ursprungsgerade verwendet? [1P]
Warum werden so niedrige Konzentrationen für den Versuch gewählt? [1P]
Flammenfärbung verschiedener Alkali-Metall-Salze
Alle Elemente emittieren im atomaren oder ionisierten gasförmigen Zustand bei hohen Temperaturen für das Element charakteristisches Licht. Bei den Alkali-, Erdalkali- und einigen anderen Elementen lässt sich dies bereits in einer Bunsenbrennerflamme erreichen.
Gegeben sind fünf Proben bekannter Alkali- und Erdalkalisalze. Betrachten Sie die Flammenfärbung der verschiedenen Substanzen. Verwenden Sie dazu ausgeglühte Magnesiastäbchen, mit denen Sie eine kleine Menge der zu untersuchenden Substanz aufnehmen und in die Flamme halten.
Zur Analyse erhalten Sie eine unbekannte Probe und sollen diese einem der fünf bekannten Kationen zuordnen.
Vergleichen Sie die Farbeindrücke der jeweiligen Kationen. [1]
Welches Kation war in der unbekannten Substanz enthalten? [1P]
Warum soll bei der Handhabung der Magnesiastäbchen Hautkontakt vermieden werden? [1P]
Vollanalyse
Konzentrationsbestimmung von Farbstofflösungen und Farbstofflösungsgemischen
Unter Verwendung des Lambert-Beerschen-Gesetzes sollen die Konzentration einer Farbstofflösung unbekannter Konzentration bestimmt werden. Außerdem werden die Konzentrationen der Komponenten eines Lösungsgemisches aus Chinolingelb und Patentblau V ermittelt. Der Extinktionskoeffizient für Chinolingelb ist aus der Übungsanalyse bekannt.
Stellen Sie eine Verdünnungsreihe für Patentblau V mit folgenden Konzentrationen her: 0,002 mmol L−1, 0,003 mmol L−1, 0,004 mmol L−1 und 0,005 mmol L−1. Das Lösungsmittel ist Wasser. Die Küvette sollte dabei für jede Verdünnungsstufe auf ein Gesamtvolumen von 2 mL aufgefüllt werden. Messen Sie die Absorptionsspektren.
Bestimmen Sie den Extinktionskoeffizienten für den Patentblau V Farbstoff wie in der Übungsanalyse. [3P]
Messen Sie nun Absorptionsspektren der Farbstofflösung unbekannter Konzentration und Zusammensetzung (Lösung A), sowie das Farbstoffgemisch (Lösung B).
Stellen Sie die Spektren der beiden Farbstofflösungen in einem Graphen dar. [1P]
Welchen Farbstoff enthielt die Lösung A? Bestimmen Sie die Konzentration mit Hilfe des Lambert-Beer-Gesetzes. [2P]
Bestimmen Sie die Konzentrationen von Chinolingelb und Patentblau V in Lösung B. Warum kann die Bestimmung über die beiden Maxima getrennt behandelt werden? [2P]
Führen Sie eine Fehlerabschätzung durch. Überlegen Sie dazu wo mögliche Fehlerquellen liegen und wie groß die jeweils resultierenden Ungenauigkeiten sind. [2P]
Chemolumineszenz – Luminol – Blutspurentest
Bei der Chemolumineszenz wird bei einer exothermen Reaktion die frei werdende Energie in Form von Licht, meist im sichtbaren Bereich zwischen 400 und 700 nm, ausgestrahlt. Da es sich um keine Temperaturstrahlung handelt, wird es oft als „kaltes Licht“ bezeichnet. Die Chemolumineszenz tritt bei vielen chemischen Reaktionen auf, in denen energiereiche, angeregte (instabile) Zwischenstufen entstehen und sofort wieder, unter Emission von Licht, zerfallen.
Die bekannteste Chemolumineszenz-Reaktion ist die Oxidation von Luminol mit Wasserstoffperoxid in Gegenwart von Eisen- oder Mangan-Ionen, die zur Sichtbarmachung von Blutspuren genutzt wird (der Blutfarbstoff Hämoglobin enthält Eisen-Ionen). Das oxidierte Luminol-Molekül dient dabei gleichzeitig als Sensibilisator. Ein weiteres Beispiel sind kommerziell erhältliche Kunststoffröhrchen, die beim Knicken ein intensives, verschiedenfarbiges, lang anhaltendes Licht abgeben. Dieses Licht wird ebenfalls durch Chemolumineszenz erzeugt. In den Röhrchen befinden sich drei Komponenten: ein Oxalsäureester, ein Farbstoff, dessen Emission die Farbe des Lichtes bestimmt, und ein Röhrchen mit Wasserstoffperoxid. Wird das Röhrchen mit Wasserstoffperoxid zerbrochen, startet die Peroxyoxalat-Chemolumineszenz.
Teil A: Stellen Sie die Lösungen I und II her.
I: Lösen Sie 0,05 g Luminol in 10 mL 10 %ige NaOH. Verdünnen Sie anschließend auf 100 mL.
II: Lösen Sie 0,25 g K3[Fe(CN)6] in 10 mL 5 %iger Wasserstoffperoxidlösung.
Lösung II muss immer frisch zubereitet werden. Geben Sie in einem dunklen Raum Lösung II zur Lösung I und notieren Sie ihre Beobachtungen.
Teil B: Gegeben ist ein Stofftuch mit zwei roten Substanzen (Fleck 1 und Fleck 2). Zur Analyse wird pro Praktikumssaal eine Lösung in einer Sprühflasche folgendermaßen hergestellt: Lösen Sie 0,1 g Luminol und 5 g Natriumcarbonat in 100 mL destilliertem Wasser. Zu dieser Lösung werden 15 mL 30 %ges Wasserstoffperoxid zugefügt. Dann wird die Lösung in eine Sprühflasche gefüllt (Achtung: ungelöste Rückstände können die Düse verstopfen). Besprühen Sie nun die roten „Flecken“ und notieren Sie ihre Beobachtungen.
Was beobachten Sie nach Zusammengeben der Lösungen I und II? [1P]
Welche Funktion hat das Kaliumhexacyanoferrat? [1P]
Welcher Fleck ist ein Blutfleck und woran erkennt man dies? [1P]
Formulieren Sie die Luminolreaktion. [1P]
Diese allgemeinen Konzepte sollten Sie hinter den Versuchen erkennen
Farbeindrücke können sehr verschiedene Ursachen haben. Hierzu gehören die Absorption, Reflexion und Streuung von Licht, die Aussendung von Licht als Fluoreszenz oder Phosphoreszenz sowie Chemolumineszenz und die hier nicht besprochene Wärmestrahlung.
Weißes Licht resultiert aus der Überlagerung von Lichtwellen unterschiedlicher Farben (Wellenlängen). Farbeindrücke entstehen wenn nur bestimmte Farben vorhanden sind oder wenn Farben „fehlen“. Die Charakterisierung einer Lichtquelle und ihres Farbeindrucks erfolgt anhand ihres Spektrums, welches den spektral abhängigen Intensitätsverlauf aufzeigt.
Sortieren Sie die Farben Rot, Blau, Gelb und Grün nach aufsteigender Wellenlänge. [1P]
Ist natürliches Licht polarisiert? [1P]
Welcher Zusammenhang besteht zwischen der Absorptions- und der Fluoreszenzwellenlänge eines Stoffes? [1P]
Elektrochemie
In der Elektrochemie werden die Grundlagen für die Entwicklungen leistungsstarker Lithiumbatterien für Mobiltelephone und PCs, für Bleiakkumulatoren in Automobilen oder Brennstoffzellen zur Umwandlung von Methanol oder Wasserstoff in elektrische Energie in U-Booten, bzw. Raumstationen gelegt. Besonders das letztgenannte Arbeitsgebiet der Brennstoffzellen-Entwicklung ist dabei gegenwärtig als interdisziplinäres Feld der Physik, Chemie und den Materialwissenschaften aktuell. Auch in biologischen Systemen spielen elektrochemische Vorgänge eine bedeutende Rolle: man denke hierbei nur an die Atmungskette, die Cytochromoxidasen, die Erregungsleitung in Nervenzellen und viele weitere Vorgänge, die mit dem Verschieben von Elektronen und Ladungen auf zellulärer Ebene einhergehen. Elektrochemie ist dabei immer auch als Teilgebiet des wesentlich größeren Feldes der Chemie der Redoxreaktionen aufzufassen. Viele formale Dinge wie die Nernst'sche Gleichung oder das Aufstellen stöchiometrischer Redoxgleichungen finden sich deshalb hier in diesem Kapitel wieder.
Wissenswertes vorab
Aus dem Physik-Unterricht der Schule ist bekannt, dass elektrischer Strom modellhaft als Ladungstransport aufgefasst werden kann. Der Transport von Ladungen ist dabei an Ladungsträger wie Kationen, Anionen oder - im einfachsten Fall - Elektronen gebunden. Elektrischer Strom benötigt prinzipiell kein leitendes Medium, sondern kann auch im Vakuum erfolgen. Nachfolgende Versuche beschränken sich jedoch nur auf Vorgänge mit Ladungstransport in festen oder flüssigen leitenden Medien.
Die Beziehung zwischen Stromstärke I (SI-Einheit: Ampere, A), Spannung U (SI-Einheit: Volt, V) und elektrischem Widerstand R (SI-Einheit: Ohm, Ω) wird durch das Ohm'sche Gesetz beschrieben:
R = U/I
Bitte beachten: aus historischen Gründen
fließt in der Sprache der Techniker der elektrische Strom vom
Pluspol einer Stromquelle zum Minuspol (Technische
Stromrichtung,
es werden also positiv geladene Ladungsträger bewegt),
wohingegen
der tatsächliche Stromfluss der Bewegung negativ geladener
Elektronen zum Pluspol einer Stromquelle entspricht (Physikalische
Stromrichtung). Im Folgenden wird nur die physikalische
Stromrichtung verwendet.
Die Beschreibung eines galvanischen Elements findet
sich im
Abschnitt "Bleiche, Desinfektion, oxidativer Stress: starke
Oxidationsmittel" des Skripts. Die folgende Abbildung zeigt als
klassisches Beispiel einer galvanischen Zelle das Daniell-Element:
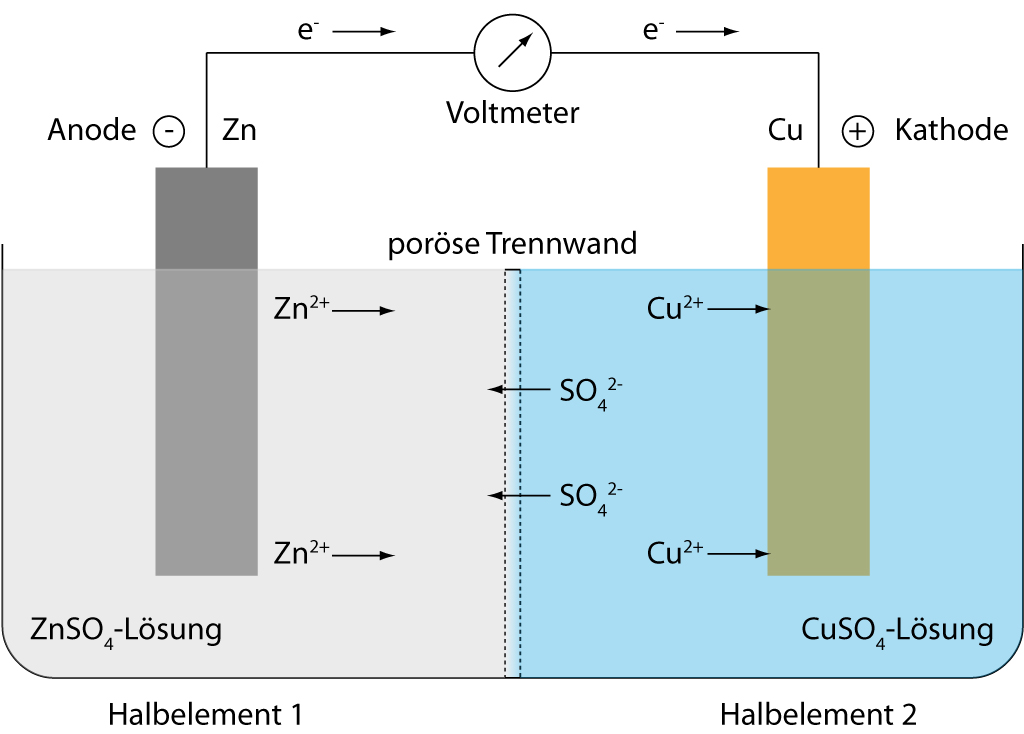
Es besteht aus einer Zinkhalbzelle die gegen eine
Kupferhalbzelle geschaltet ist. Bei einem galvanischen Element ist die Anode
der negative Pol an dem im Beispiel des Daniell-Elements elementares Zn
zu Zn2+ oxidiert wird. Die Kathode
ist der positive Pol an dem die Kupferionen zu elementarem Kupfer
reduziert werden.
Es muss zwischen galvanischen Zellen und Elektrolysezellen
unterschieden werden. Galvanische Zellen wandeln durch freiwillig
ablaufende Reaktionen chemische Energie in elektrische Energie um. Bei
Elektrolysezellen wird elektrische Energie zugeführt, um eine
nicht freiwillig ablaufende chemische Reaktion in der Zelle zu
erzwingen. Hierbei ist die Polung der Elektroden invers zur Polung in
einer galvanischen Zelle. Die Anode ist der positive, die Kathode der
negative Pol.
Aus dem Alltag sind galvanische Zellen als Batterien bekannt. Ist der
Entladevorgang einer galvanischen Zelle durch Energiezufuhr reversibel
spricht man von einem Akkumulator (Akku). Folglich können die
Vorgänge beim Entladen eines Akkumulators als galvanische
Zelle,
beim Laden als Elektrolysezelle beschrieben werden.
Lernziele und einführende Literatur
Lernziele: Einschätzung der Leitfähigkeit wässriger Lösungen in Gegenwart bestimmter Ionen, Bestimmung der Polung einer Gleichstromquelle, elektrochemische Spannungsreihe und Konzentrationsgradienten als Ursache für Potentialgefälle, Bleiakkumulator, Maßanalytische Bestimmung am Beispiel der Konduktometrie
Einführende Literatur: Mortimer (Kapitel 20), Atkins (Kapitel 11 und 12)
Einführende Versuche
Die ersten Versuche sollen Ihnen ein Gefühl dafür vermitteln, welche Zusammenhänge zwischen Ladungstransport, Ladungsträgern und dem Stromfluss an sich sowie den dahinterliegenden und einhergehenden chemischen Vorgängen besteht. Nehmen Sie sich für die Versuche genügend Zeit! Da Messungen stets einen gewissen statistischen Fehler mit sich bringen, sollten Messungen mehrere Male durchgeführt werden, um diesen - zumindest ansatzweise - auszugleichen.
Leitfähigkeit von Ionen in wässriger Lösung
Lernziel: Sie sollen ein Gefühl dafür entwickeln, in welchem Umfang sich die Leitfähigkeit wässriger Lösungen in Abhängigkeit der Konzentration und Natur anwesender Ionen verändert.
Jede einzelne der hierzu benötigten wässrigen
Salzlösungen soll von jeweils zwei Praktikanten angesetzt
werden
und kann dann im Tausch von allen Kursteilnehmern im Saal verwendet
werden. Die Lösungen sollten bereits am Vortag bzw. in der
Vorwoche hergestellt werden, um die Einstellung der Lösungen
auf
Zimmertemperatur sicherzustellen. Mittels der Messkolben (250 mL)
werden jeweils 0,1 m
Lösungen der Salze aus der untenstehenden Tabelle hergestellt
(beachten Sie bei Ihren Berechnungen, dass einige der Salze in
kristalliner Form als definierte Hydrate vorliegen).
Ferner sollen ausgehend von konzentrierter Salzsäure bzw.
festem Natriumhydroxid 0,1 m
Lösungen von Salzsäure und Natronlauge hergestellt
werden.
Der Versuchsaufbau besteht aus einem Konduktometer (siehe Bild) mit
einer Leitfähigkeitselektrode.
Alle Salzlösungen sollen auf ihre jeweiligen
Leitfähigkeiten
hin überprüft werden.
Zweckmäßigerweise geht man
hierzu folgendermaßen vor: das Konduktometer wird
eingeschaltet
und vor der ersten Verwendung mit den bereitgestellten
Kalibrierlösungen kalibriert. Die
Leitfähigkeitselektrode
wird in die zu messende Flüssigkeit eingetaucht und der
angezeigte
Wert abgelesen. Vor jeder Messung müssen die Elektroden
gründlich mit deionisiertem Wasser abgespült werden.
Als
letztes soll die Leitfähigkeit der Natronlauge und der
Salzsäure bestimmt werden.
| K+ | K2+ | A- | A2- | |
|---|---|---|---|---|
| LiCl | MgCl2 | NaF | Na2SO4 | NaOH |
| NaCl | BaCl2 | NaI | Na2C2O4 | HCl |
| KCl |
Starke und schwache Elektrolyte Kx+Ay- mit unterschiedlicher Ionenstärke.

Tragen Sie die ermittelten Stromstärken tabellarisch auf und
diskutieren Sie die Änderung der Leitfähigkeit in der
Reihe
der einwertigen und zweiwertigen Kationen sowie der einwertigen und
zweiwertigen Anionen. [3P]
Erklären Sie die überraschend gute
Leitfähigkeit der
Natronlauge und der Salzsäure im Vergleich zur
ungesättigten
Natriumchlorid-Lösung. [2P]
Welche Veränderung der Leitfähigkeit würden
Sie bei
einer gesättigten Salzlösungen erwarten?
Begründen Sie
Ihre Annahmen. [2P]
Bestimmung der Polung einer Gleichstromquelle
Lernziel: Stromfluss durch wässrige Lösungen ist in vielen Fällen mit einer Veränderung des pH-Werts verbunden. In einer hierfür geeigneten Versuchsanordnung kann dieser Effekt zur Bestimmung der Polung einer Gleichstromquelle (z.B. einer Batterie) benutzt werden.
Die beiden Pole einer 4,5-Volt-Batterie werden jeweils mit einem Draht
verlängert (welche zuvor gründlich mit Wasser und
Seife
gereinigt wurden). Anschließend wird eine etwa 0,1 m
wässrige Lösung von Natriumchlorid hergestellt und
mit
einigen Tropfen Phenolphthalein-Lösung versetzt. Mit dieser
wird
ein Streifen Filterpapier (etwa 5 × 1 cm) getränkt,
welcher
danach auf einem Uhrglas ausgebreitet wird.
Vorsicht: der Filterpapierstreifen soll für dieses
Experiment
zwar feucht sein, auf dem Uhrglas soll jedoch keine Pfütze
stehen!
Die Enden der beiden Drähte werden in einem Abstand von etwa 2
cm nebeneinander auf den Filterpapiertstreifen gepresst.
Notieren und erklären Sie alle Beobachtungen und formulieren
Sie
die ablaufenden Reaktionen. Wie kann daraus eindeutig die Polung der
Batterie abgeleitet werden? [2P]
Wiederholen Sie den Versuch mit einem neuen Filterpapierstreifen und stellen Sie dieses Mal einen Abstand von 4 cm zwischen den beiden Drähten auf dem Filterpapier ein.
Wie ist der zeitlich andere Verlauf der Beobachtungen zu
erklären? [1P]
Galvanische Zellen
Lernziel: Wie wird in einer Batterie überhaupt elektrischer Strom erzeugt? Sie sollen erkennen, dass durch den Kontakt verschiedener Metalle und Metallionen Potentialgefälle erzeugt werden. Das Ausmaß des Potentialgefälles hängt dabei sowohl von der Konzentration der beteiligten Reaktionspartner als auch von der unterschiedlichen Stellung der Reaktanden in der elektrochemischen Spannungsreihe ab.
Bleinitrat ist als fruchtschädigend eingestuft. Schwangere sollten daher in keinem Fall Umgang mit Bleinitrat haben. (Kann das Kind im Mutterleib schädigen. Kann möglicherweise die Fortpflanzungsfähigkeit beeinträchtigen. R-Sätze: R61-20/22-33-50/53-62. Bitte beachten Sie auch das Sicherheitsdatenblatt).
Zunächst werden 0,1 m
wässrige
Lösungen von Bleinitrat, Zinknitrat und Kupfernitrat
vorbereitet
und in Bechergläser (100 mL) gefüllt.
Anschließend
werden jeweils zwei Streifen Kupferblech, Zinkblech und Bleiblech
gründlich mit Wasser und Seife gereinigt (Vorsicht vor
scharfen
Kanten!), mit einigen Tropfen Salpetersäure (6 M) und
Zellstoff
von anhaftenden Verunreinigungen befreit, erneut gründlich mit
Wasser gespült (blankes, glänzendes Metall muss
sichtbar
sein!) und in die zugehörige Salzlösung gestellt.
Diese
Anordnung wird jeweils als „Halbelement“ bezeichnet.
Aus den bereitgestellten U-förmig gebogenen Glasrohren werden
jetzt Salzbrücken gebaut, indem man diese vollständig
mit
wässriger 0,5 m
Ammoniumnitrat-Lösung füllt und an beiden Enden mit
einem
Wattepfropf luftblasenfrei verschließt. Man verwendet
zunächst die Glasrohre mit einem mittleren Innendurchmesser
(etwa
7 mm).
Zur Messung der sich zwischen zwei Halbelementen ausbildenden Spannung
verbindet man die Metallblechstreifen zweier nebeneinanderstehender
Halbelemente mit einem Voltmeter (Krokodilklemmen verwenden), verbindet
die beiden Halbelemente durch Eintauchen der Salzbrücke und
liest sofort
die Spannung ab. Nach der Messung wird die Salzbrücke wieder
aus
den beiden Halbelementen herausgezogen. Um eine Messung mehrfach zu
wiederholen, merkt man sich, welches der Enden mit welcher
Salzlösung in Kontakt stand, da diese beim neuerlichen
Eintauchen
keinesfalls vertauscht werden dürfen.
Folgende Teilexperimente sollen in der angegebenen Reihenfolge
durchgeführt werden:
• Spannungsmessung unter Verwendung unterschiedlicher
Elektrodenpaare (Pb/Zn, Pb/Cu und Cu/Zn; s. a. Frage 3.6 und 3.7)
• Spannungsmessung unter Verwendung von Salzbrücken
mit unterschiedlichem Innendurchmesser (s. a. Frage 3.8)
• Spannungsmessung mit Cu/Cu Element in zwei 0.1 m Cu(NO3)2
Lösungen (s. a.
Frage 3.9)
• Spannungsmessung mit Cu/Cu Element in 0.1 m und 0.01 m
Cu(NO3)2
Lösung (s. a.
Frage 3.10, Teil 1). Spannungsmessung mit Cu/Cu Element in 0.1
m und 0.001 m
Cu(NO3)2
Lösung (s. a.
Frage 3.10, Teil 2).
Was folgt aus den erhaltenen Werten der Spannungsmessung der Paare
Pb/Zn und Pb/Cu über die Einordnung der drei Metalle relativ
zueinander in der elektrochemischen Spannungsreihe? Welche Spannung
kann man deshalb für das Paar Cu/Zn erwarten? Wird dieser Wert
auch experimentell erhalten? [2P]
Vergleichen Sie Ihre Messergebnisse mit den erwarteten Werten, die sich
aus tabellierten Normalpotentialen ergeben hätten. Was
könnten die Ursachen für Abweichungen sein? [2P]
Wie ändern sich die Unterschiede zwischen experimentellen und
theoretischen Werten, wenn Salzbrücken mit kleinerem und
größerem Innendurchmesser verwendet werden? Wie
könnte
man folglich die Unterschiede noch weiter minimieren? Was wäre
anstelle einer Salzbrücke idealerweise zu verwenden? [3P]
Ersetzen Sie nun im Cu/Zn-Paar den Zinkblechstreifen durch einen
Kupferblechstreifen, wodurch der Cu-Elektrode jetzt eine weitere
Cu-Elektroden gegenübersteht. Welche Spannung wird jetzt
zwischen
den beiden Halbelementen gemessen? Was für eine Anordnung
liegt
jetzt vor? Wie könnte man die Spannung dieser Cu/Cu-Zelle noch
weiter erhöhen? [3P]
Überprüfen Sie experimentell durch Herstellung einer
1:10 und 1:100 Verdünnung der Cu(NO3)2
Lösung die
resultierende Zellspannung der Cu | Cu(NO3)2
(0,01 m) || Cu(NO3)2
(0,1 m)| Cu Zelle
sowie der Cu | Cu(NO3)2(0,001
m) || Cu(NO3)2
(0,1 m) | Cu Zelle. Um welchen Wert sollte
sich die Zellspannung der verschiedenen Verdünnungnen jeweils
theoretisch unterscheiden (Hinweis zur Herstellung der
Verdünnungen: Nach Messung der Spannung in Aufgabe 4.9
entnehmen
Sie 5 mL der 0.1 m Cu(NO3)2 Lösung
und geben diese in ein frisches Becherglas. Fügen Sie 45 mL H2O
(VE) hinzu um eine 1:10 Verdünnung zu erhalten. Zur
Herstellung
der 1:100 Verdünnung gehen Sie analog vor und entnehmen
entsprechend 5 mL der 1:10 Verdünnung und geben diese wieder
in
ein frisches Becherglas und geben auch hier 45 mL H2O
(VE) hinzu. Hinweis zur Theorie: Nernst-Gleichung)? [3P]
Vollanalyse Elektrogravimetrie
Die Elektrolyse von Metallsalzlösungen und -schmelzen stellt ein wichtiges Reinigungsverfahren für Metalle und in einigen Fällen die Hauptdarstellungsmethode elementarer Metalle dar. Mittels elektrolytischer Verfahren kann auch der Gehalt einer Lösung an Metall-Ionen gravimetrisch bestimmt werden. Besonders gut verläuft diese Bestimmung bei edleren Metallen wie Kupfer, deren Ionen sich leicht an einer negativ geladen Elektrode abscheiden lassen.
Die Versuchsanordnung besteht aus einer Elektrolysestation und zwei
Platinelektroden in Form eines Drahtnetzes und eines
spiralförmig
gebogenen Drahtes. Zunächst sollten Sie sich mit der
Funktionsweise der
Elektrolysestation vertraut machen und die Polung der Gleichstromquelle
überprüfen (das Rüstzeug dazu haben Sie in
einem der Vorversuche
erhalten). Machen Sie sich klar, an welchem Pol das Drahtnetz und an
welchem Pol die Platinspirale angeschlossen werden muss.
Das Platinnetz und der Platindraht werden zunächst mit
heißer konzentrierter Salpetersäure, welche keine
Spuren von Chlorid enthalten darf,
gereinigt. Die Säure wird dazu am besten zentral für
alle anwesenden
Praktikanten in einem größeren Becherglas auf etwa
100 °C erhitzt und
die Platinelektroden werden für etwa eine Minute eingetaucht.
Anschließend spült man der Reihe nach sehr
gründlich mit deionisiertem
Wasser und absolutem Alkohol (Achtung!
Nicht über der konzentrierte Salpetersäure
spülen und
danach auf keinen Fall wieder in die Säure eintauchen!)
und trocknet bei 110 °C im Trockenschrank für etwa 30
Minuten. Nach dem
Abkühlen auf Raumtemperatur bestimmt man an der Analysenwaage
die Masse
der Drahtnetzelektrode. Anschließend wird der
spiralförmige Platindraht
als Anode, die Elektrode in Form des Drahtnetzes als Kathode in das
Elektrolysegerät eingebaut. Der spiralförmige Draht
wird dabei
vollständig vom Netz umgeben, darf dieses jedoch keinesfalls
berühren
(auch beim späteren Rühren nicht!).
Ein Aliquot (50 mL) der auf die übliche Weise vorbereiteten
Analysenlösung wird in ein hohes Becherglas (150 mL) gegeben,
mit 10 mL
Schwefelsäure (20%) versetzt und auf ein Gesamtvolumen von
70–80 mL
verdünnt. Die Elektroden sollen dabei nur etwa zur
Hälfte in die
Probenlösung eintauchen. Man erwärmt den Inhalt des
Becherglases unter
Rühren mit einem Rührfisch auf 70°C und
elektrolysiert bei einer
Spannung von 2,5–3,0 Volt. Nach etwa 20 Minuten
erhöht man den
Flüssigkeitsstand etwas durch Zugabe von deionisiertem Wasser
und
überprüft, ob sich an der jetzt neu in die
Lösung eintauchenden Fläche
im Laufe einiger Minuten noch etwas Kupfer abscheidet. Der Vorgang wird
ggf. solange wiederholt, bis sich auch 15 Minuten nach Zugabe von
deionisiertem Wasser unter fortgesetzter Elektrolyse keine
Kupferabscheidung mehr beobachten lässt.
Nach beendeter Elektrolyse werden die Elektroden aus der
Probenlösung gezogen und (noch unter Spannung) mit
deionisiertem Wasser
abgespült. Anschließend wäscht man diese
noch mit reinem Alkohol,
trocknet im Trockenschrank bis zur Gewichtskonstanz und bestimmt erneut
die Masse.
Geben Sie den Gehalt an Kupfer in Ihrer Maßlösung
an. [1P]
Bis zu welcher Restkonzentration kann man die Kupfer-Abscheidung aus
saurer Lösung (pH = 1) theoretisch beobachten, bevor an der
Kathode Wasserstoff entwickelt wird? Geben Sie Ihren Rechenweg an (E0(Cu/Cu2+)
= 0,345 V) [3P]
Wie groß war die mittlere Stromstärke (Ampere, A),
wenn die vollständige Abscheidung von Cu-Ionen in
Lösung innerhalb von 30 min erfolgte? [2P] Wie viel Ladung
(Coulomb, C) ist in diesem Zeitraum geflossen? [1P]
Diese allgemeinen Konzepte sollte Sie hinter den Versuchen erkennen
Bei einer galvanischen Zelle läuft die chemische
Reaktion
freiwillig ab und dies kann genutzt werden um eine Spannung zu
erzeugen. Hingegen muss bei einer Elektrolysezelle Spannung angelegt
werden, damit eine nicht freiwillig ablaufende Reaktion stattfindet.
Die Konzentrationsabhängigkeit des Potentials lässt
sich mit der Nernstgleichung bestimmen.
Anorganische Chemie
Sie haben im ersten Teil des Praktikums neben
grundlegenden Arbeitstechniken auch den sicheren Umgang mit
Gefahrstoffen geübt sowie die Bedeutung von Betriebsanweisungen
kennengelernt. Dieses Wissen, dass chemische Substanzen in der Regel
Gefahrstoffe sind und einen besonnenen Umgang mit ihnen benötigen,
"dürfen" Sie nicht nur, sondern müssen Sie jetzt auch
umsetzen!
Das Protokoll zum
Anorganisch-chemischen Teil des Praktikums können Sie hier als
pdf-Version
herunterladen.
Reaktionskinetik
Die Geschwindigkeit mit der eine Reaktion abläuft, kann von Reaktion zu Reaktion stark variieren. So laufen die Explosion von Nitroglycerin oder die Neutralisation von NaOH und HCl innerhalb von Bruchteilen einer Sekunde ab. Die Faltung eines Proteins kann dagegen abhängig von der Größe einige Sekunden bis Minuten dauern, das Rosten von Eisen benötigt schon einige Jahre und die Umwandlung von Diamant zu Graphit verläuft nicht messbar langsam (findet jedoch statt). Bei vielen Reaktionen ist es jedoch wichtig eine quantitative Aussage zur Reaktionsgeschwindigkeit machen zu können. So ist es zum Beispiel in der Pharmakologie wichtig die Wirkungsdauer eines Medikamentes, in der Biologie die Dauer einer enzymatischen Reaktion oder den radioaktiven Zerfall von natürlich vorkommenden Isotopen bei der Altersbestimmung von Gegenständen (zum Beispiel der Zerfall von 14C bei der Radiocarbonmethode) zu bestimmen. Die Reaktionskinetik beschreibt mit mathematischen Ausdrücken die Konzentrationsänderung der Reaktanden und der Produkte in Abhängigkeit von der Zeit und somit mit welcher Geschwindigkeit eine Reaktion abläuft.
Wissenswertes vorab
Betrachtet wird eine Reaktion der Form
A → B
[A] und [B] bezeichnen die Konzentrationen der Reaktanden A und B zu einem beliebigen Zeitpunkt t.
Reaktionsgeschwindigkeit: Die Reaktionsgeschwindigkeit v kann als Bildungsgeschwindigkeit des Produkts B
vB = d[B]/dt
oder als Verbrauchsgeschwindigkeit des Edukts A
vA = -d[A]/dt
angegeben werden.
Geschwindigkeitsgesetz: Die Reaktionsgeschwindigkeit einer Reaktion ist gewöhnlich proportional zu irgendeiner Potenz der Konzentrationen der Reaktanden. Für eine Beispielreaktion etwa von drei Reaktanden A, B und C könnte gelten:
v = k·[A]x·[B]y·[C]z
Die Proportionalitätskonstante k wird als Geschwindigkeitskonstante bezeichnet. Sie ist nicht abhängig von der Konzentration der Reaktanden, hängt aber von der Temperatur T ab. x, y und z geben die Reaktionsordnung an. Die Gesamtreaktionsordnung ergibt sich aus deren Summe.
Das Geschwindigkeitsgesetz ist mathematisch betrachtet eine Differentialgleichung. Durch Integration des Geschwindigkeitsgesetzes wird das integrierte Zeitgesetz erhalten. Aus dem Zeitgesetz der Reaktion und der Lösung der Differentialgleichung, kann die Konzentration eines beliebigen Reaktanden zu einem beliebigen Zeitpunkt vorhergesagt werden.
Reaktionsordnung: Die gesamte Reaktionsordnung entspricht der Summe der Potenzen, mit der die jeweiligen Reaktanden im Geschwindigkeitsgesetz erscheinen. Die Reaktionsordnung muss nicht ganzzahlig sein. Wichtige Spezialfälle sind jedoch Reaktionen 0., 1. und 2. Ordnung.
Übersicht über die einfachsten Reaktionsordnungen
| 0. Ordnung | 1. Ordnung | 2. Ordnung | |
|---|---|---|---|
| Typ | A + Katalysator → B + C + Katalysator | A → B + C | 2A → C + D |
| Geschwindigkeitsgesetz | -d[A]/dt = k0 | -d[A]/dt = k1·[A] | -d[A]/dt = k2·[A]2 |
| integriertes Zeitgesetz | [A] = -k0·t + [A]0 | [A] = [A]0·exp(-k1·t) | 1/[A] = k2·t + 1/[A]0 |
| Halbwertszeiten | (t½)0 = 1/(2·k0)·[A]0 | (t½)1 = ln(2)/k1 | (t½)2 = 1/k2·1/[A]0 |
| Beispielreaktion | von Proteinen katalysierte Reaktionen in einer Zelle | Radioaktiver Zerfall | H2 + I2 → 2 HI oder die Dimerisierung von Butadien in der Gasphase |
ACHTUNG:
Aus der Reaktionsgleichung kann nicht direkt auf die Reaktionsordnung geschlossen werden!
Betrachtet wird die Reaktion:
H2 + Br2 → 2 HBr
Diese ist nicht zwangsweise 2. Ordnung, obwohl dies auf dem Papier zunächst so erscheinen mag!
Reaktionen dieses Typs können zweiter Ordnung sein und es fällt uns leicht dies mit der Reaktionsgleichung in Einklang zu bringen. Wir stellen uns vor, die Edukte A und B stoßen zusammen und reagieren zu den Produkten C (und ggf. D).
Die Reaktionsordnung hängt allerdings vom Reaktionsmechanismus einer Reaktion ab. Dieser ist aus der Reaktionsgleichung nicht immer ersichtlich. Die obenstehende Reaktion verfügt über eine einfache Stöchiometrie, allerdings über einen komplizierten Reaktionsmechanismus. So kann es etwa sein, dass nicht die Edukte selbst miteinander reagieren, sondern erst reaktive Zwischenprodukte aus den Edukten entstehen müssen, die dann miteinander zu den Produkten reagieren.
Deshalb kann für die obenstehende Reaktion bei manchen Versuchsbedingungen überhaupt keine Gesamtreaktionsordnung angegeben werden.
In der Praxis muss durch Versuchsreihen der Reaktionsmechanismus aufgeklärt und anschließend auf die Reaktionsordnung zurückgeschlossen werden.
Lernziele und einführende Literatur
Lernziele: Beschreibung der Reaktionskinetik durch mathematische Formeln, Erkennen der Reaktionsordnung durch graphische Auswertung der Messergebnisse
Einführende Literatur: Charles E. Mortimer Chemie, 9. Auflage 2007 (Kapitel 15), Peter W. Atkins, Julio de Paula Physikalische Chemie, 4. Auflage 2006 (Kapitel 22), Peter W. Atkins, Julio de Paula Kurzlehrbuch Physikalische Chemie, 4. Auflage 2008 (Kapitel 15)
Vorversuche
Beobachtung von unterschiedlich schnell ablaufenden Reaktionen.
Reaktion von Kristallviolett
Kristallviolett ist ein organischer Farbstoff aus der Klasse der Triphenylmethyl-Farbstoffe. Das Molekül kann bei Bestrahlung
Licht einer
bestimmten Wellenlänge aus dem sichtbaren Bereich des elektromagnetischen Spektrums absorbieren.
Wir nehmen schließlich die Komplementärfarbe des
absorbierten Lichts als Farbeindruck wahr. Im Fall des Kristallvioletts bedeutet das: Die Substanz absorbiert das gelbe Licht
und sie erscheint deshalb violett.
Die Farbigkeit des Moleküls basiert auf einem über das ganze Molekül delokalisierten π-Elektronensystem. Durch die
Delokalisierung wird das Absorptionsmaximum des Farbstoffs in den sichtbaren Bereich des elektromagnetischen Spektrums
verschoben. Die Delokalisierung findet jedoch nur statt,
wenn das Molekül eine planare Geometrie, ähnlich einer Scheibe, aufweist. Die planare Struktur und das delokalisierte System
gehen verloren, wenn eine Base, z.B. KOH, am zentralen Kohlenstoffatom nukleophil angreift und sich so eine Hydroxylgruppe (-OH)
anlagert. Das Molekül absorbiert nun andere Wellenlängen aus dem nicht sichtbaren Bereich des Lichtspektrums und die Substanz
erscheint farblos.
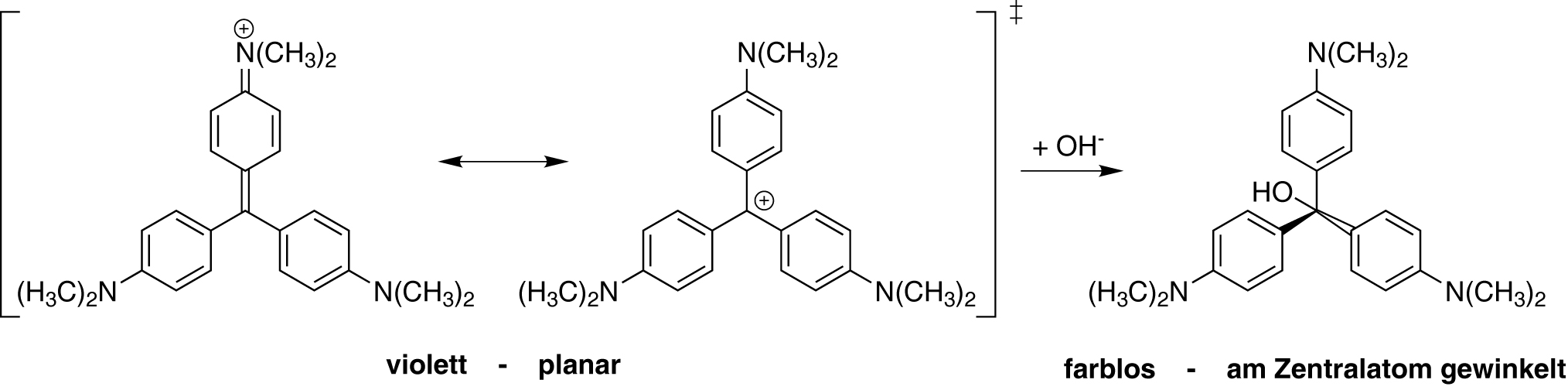
In einem Uhrglas werden 1 Tropfen Kristallviolettlösung aus dem Indikatorsatz und 4 mL Wasser miteinander vermischt. In einem
zweiten Uhrglas werden 1 mL Kristallviolett mit 4 mL 3 m KOH gegeben.
Beschreiben Sie Ihre Beobachtung. [1P]
In welchem Wellenlängenbereich des Spektrums liegt das Absorptionsmaximum vor und nach der Anlagerung des Hydroxidions?
(Wellenlängenbereich angeben!) [2P]
Wieso kann die Reaktion auch mit Carbonaten in wässriger Lösung durchgeführt werden? (Reaktionsgleichung angeben!) [2P]
Wie ist das Zentralatom vor und nach der Anlagerung der Base hybridisiert? Wie groß sind jeweils die Bindungswinkel am Zentralatom? [2P]
Vollanalyse Fluoreszenz
Kinetik der Lumineszenz von Leuchtsternen
Leuchtsterne in Kinderzimmern leuchten noch einige Zeit, nachdem das Licht gelöscht wurde. Der folgende Versuch beschäftigt sich mit der Chemie, die hinter den Leuchtsternen steckt. Die Leuchtsterne bestehen aus Zinksulfid, ZnS. ZnS ist namensgebend für den Zinkblende-Strukturtyp. Die Sulfid-Anionen (S2−) bilden eine kubisch flächenzentrierte Kugelpackung (fcc), die kleineren Zn2+ Kationen besetzen die Hälfte der Tetraederlücken (1/2 TL). Außerdem ist Cu+ in sehr kleinen Konzentrationen eingelagert. Die Oxidationsstufe +I für Kupfer ist sehr selten. Für gewöhnlich bevorzugt Kupfer die stabilere Oxidationsstufe +II. Zwischen den besetzten Orbitalen des ZnS, die das Valenzband (VB) bilden und den unbesetzten (dem Leitungsband, LB) befindet sich eine Bandlücke, in der keine erlaubten Zustände für das ZnS vorhanden sind. Die Orbitale der eingelagerten Kupferionen befinden sich hier nun genau in der Bandlücke des ZnS, sie besetzen die lokalisierten Zustände A. Durch Bestrahlung mit Licht kann ein Elektron aus dem besetzten Valenzband in das unbesetzte Leitungsband angehoben werden (erstes Bild). Das angeregte Elektron hinterlässt so ein positiv geladenes Loch im Valenzband, da nun eine Elektronenladung zu wenig im Valenzband vorhanden ist (zweites Bild). Dieses Ladungsdefizit kann nun durch ein Elektron eines Cu+ kompensiert werden, das ein Elektron an das Valenzband des ZnS abgibt und somit selbst zu Cu2+ oxidiert wird (zweites und drittes Bild).
Die Lebenszeit des Elektrons im angeregten Zustand liegt typischerweise im ns-Bereich (= 10−9 s), d.h. die angeregten Elektronen werden innerhalb weniger ns wieder ins Valenzband zurückfallen. Dennoch kann durch permanentes Einstrahlen eine gewisse Anzahl von Löchern im Valenzband im zeitlichen Mittel gehalten werden, es wird nach einigen Sekunden eine Sättigung erreicht.
Nun wird die Bestrahlung gestoppt. Es befindet sich eine bestimmte Anzahl von Elektronen im Leitungsband und exakt die gleiche Anzahl an Löchern im Valenzband, da jedes angeregte Elektron genau ein Loch im Valenzband produziert hat. Die Reaktion des Cu+ verläuft relativ langsam, da sie thermisch aktiviert ist. Dennoch kann sie bei einer ausreichend großen Anzahl von Löchern im VB stattfinden. Somit füllen die Kupferionen mit ihren Elektronen die Löcher im Valenzband auf. Wenn eines der angeregten Elektronen im Leitungsband nun relaxiert, kann es nicht mehr in das Valenzband des ZnS zurückfallen, da das Loch bereits durch ein Elektron eines Kupfers aufgefüllt wurde. Folglich fällt das Elektron in einen Zustand des neugebildeten Cu2+ zurück und reduziert dieses wieder zu Cu+. Die überschüssige Energie wird beim Zurückfallen als Strahlung abgegeben (drittes Bild). Da die Orbitale des Kupfers energetisch oberhalb der des ZnS-Valenzbandes liegen, verkleinert sich die frei werdende Energie und damit vergrößert sich die Wellenlänge der emittierten Strahlung. Wäre das Elektron in das Valenzband zurückgefallen, wäre Strahlung im UV-Bereich des EM-Spektrums emittiert worden, die für uns nicht sichtbar ist. Die nun emittierte Strahlung liegt im sichtbaren Bereich und wird als grün-gelbe Lumineszenz wahrgenommen. Hier wird die Verweildauer des Elektrons im angeregten Zustand um einige Größenordnungen verlängert, da das angeregte Elektron ein Cu2+ benötigt, um relaxieren zu können. Da die Konzentration der Cu+ Ionen sehr gering ist, ist die Lumineszenz noch einige Minuten nach Löschen des Lichts beobachtbar (in Dunkelheit sogar einige Tage).
Beachten Sie den Unterschied zum Versuch mit Kristallviolett: Der organische Farbstoff absorbiert Licht einer
bestimmten Wellenlänge des sichtbaren Bereichs (VIS-Bereich). Der Farbstoff „subtrahiert“ mit anderen Worten also eine
Wellenlänge aus dem Weißlicht,
wir nehmen die jeweilige Komplementärfarbe der absorbierten Wellenlänge wahr.
Hier wird hingegen von den Leuchtsternen Licht einer wohldefinierten Wellenlänge im sichtbaren Bereichs emittiert, d.h. ausgesandt.
Wir nehmen direkt die Wellenlänge des emittierten Lichts wahr.
Zur Durchführung des Versuchs wird eine spezielle Küvette verwendet. In einer Aussparung der Küvette ist Kupfer dotiertes Zinksulfid eingebracht.
Die Küvette wird in den Probenhalter des Spektrometers gestellt. Es soll die
emittierte Strahlung gemessen werden, daher muss der Detektor im 90°-Winkel aufgestellt werden.
Es wird eine Messung vorbereitet, wie dies im Abschnitt Aufnahme von Emissionsspektren beschrieben wird.
Zunächst muss das Fluoreszenzmaximum der Substanz bestimmt werden. Hierfür wird ein Fluoreszenzspektrum aufgenommen. Zur Aufnahme des Fluoreszenzspektrums wird der Haken bei Strobe/Lamp Enable entfernt und die Messung durch Klicken auf Play gestartet. Durch Setzen des Hakens bei Strobe/Lamp Enable wird nun die Probe für einige Sekunden mit Licht bestrahlt. Anschließend wird der Haken bei Strobe/Lamp Enable wieder entfernt. Jetzt kann das Nachleuchten der Substanz durch Klicken auf die Zoomwerkzeuge gut beobachtet werden.
Im Anschluss wird ein Trenddiagramm im Bereich +/- 5 nm um das Fluoreszenzmaximum vorbereitet, aber noch nicht gestartet (siehe dazu den Abschnitt Aufnahme eines Trenddiagramms). Die Messung wird durch Klicken auf den oberen Start-Button gestartet. Durch Setzen des Hakens bei Strobe/Lamp Enable wird die Probe ca. 5 Sekunden mit Licht bestrahlt. Der Haken bei Strobe/Lamp Enable wird wieder entfernt und sehr schnell durch Klicken auf den unteren (kleineren) Start-Button die Messung des Trenddiagramms gestartet. Es wird solange gemessen, bis die Fluoreszenz auf das Level des Hintergrundrauschens abgefallen ist. Zu Beginn des Versuchs sollte idealerweise mindestens eine Intensität von ~ 200 Counts gemessen werden, sodass ein Abfall der Intensität klar beobachtbar ist.
Notieren Sie die im Experiment verwendete Integrationszeit.
Die Auswertung wird mit dem Programm QtiPlot durchgeführt. Dazu müssen die Messdaten exportiert und in QtiPlot eingelesen werden. Der Header der Ausgabedatei wird entfernt. Die erste Spalte beinhaltet lediglich die Uhrzeit der Messung und wird gelöscht. Die beiden verbliebenen Spalten beinhalten die Zeit in Sekunden, die nach Start der Messung vergangen sind und den jeweils zugeordneten Messwert.
Stellen Sie das Fluoreszenzspektrum des Zinksulfids in einem Schaubild dar. [2P]
Rechnen Sie die detektierte Intensität des Trenddiagramms (nicht die des Fluoreszenzspektrums!) in Intensitätswerte pro Sekunde um. Hierzu benötigen Sie die im Experiment verwendete Integrationszeit.
Beim Experiment werden Counts detektiert. Diese sollen in eine Anzahl von Photonen umgerechnet werden, da Photonen im Gegensatz zu den Counts eine anschauliche Bedeutung
als sichtbare Lichtquanten haben. Dem Datenblatt des Detektors wurde entnommen, dass 1 Count 60 Photonen entsprechen. [2P]
Verwenden Sie im weiteren Verlauf der Auswertung stets die Intensitätswerte des Trenddiagramms in Photonen/s
Werten Sie die Versuchsergebnisse des Trenddiagramms graphisch aus. Verwenden Sie die in der obigen Frage berechneten Werte für die Intensität in Photonen/s.
Welche Reaktionsordnung liegt bezüglich der Abklingreaktion vor? (welche Methode wenden Sie an, um die Reaktionsordnung zu bestimmen?)
Fitten Sie die Messdaten an und bestimmen Sie die Geschwindigkeitskonstante k der Abklingreaktion. Welche Einheit hat k? [5P]
Wie lässt sich die gefundene Reaktionsordnung mit dem im obenstehenden Schema erklärten Reaktionsmechanismus in Einklang bringen? [2P]
Das menschliche Auge nimmt in einem dunklen Raum eine Intensität von 60 Photonen/s noch als konstantes Leuchten wahr. Ein gelegentliches Aufleuchten der Substanz kann noch bis
zu einer Intensität von 5 Photonen/s wahrgenommen werden. Berechnen Sie, wie lange Sie das Nachleuchten des Zinksulfids theoretisch in einem abgedunkelten Raum als konstantes
Leuchten bzw. als gelegentliches Aufleuchten beobachten können.
Benutzen Sie hierfür Ihre Messdaten und das integrierte Zeitgesetz für die von Ihnen zuvor bestimmte Reaktionsordnung. [4P]
Wenn Sie den Versuch mit einer Lichtquelle wiederholen würden, die ausschließlich rotes Licht emittiert: Welchen Ausgang des Experiments erwarten Sie und wie lässt sich die Beobachtung erklären? [2P]
Wie sind Fluoreszenz und Phosphoreszenz definiert? Würden Sie nach den von Ihnen gefundenen Definitionen das Nachleuchten des
Zinksulfids als Fluoreszenz oder Phosphoreszenz bezeichnen? [2P]
Vollanalyse Kristallviolett
Wenn sich eine Studentin oder ein Student mit dem Füller verschreibt, greift sie oder er meist zu einem Tintenkiller. Im nachfolgenden Versuch soll verstanden werden, welche Chemie hinter einem Tintenkiller steckt. Tinte besteht unter anderem aus einem organischen Farbstoff, meist einem Triphenylmethyl-Farbstoff. Der Tintenkiller enthält reduzierende Inhaltsstoffe wie Sulfite, Carbonate oder Thiosulfate, die das zentrale Kohlenstoffatom des Farbstoffes reduzieren. Dadurch wird die planare Geometrie zerstört und das farbgebende π-System geht verloren. Folglich entfernt ein Tintenkiller die Tinte nicht, sondern entfärbt den Farbstoff nur. Im folgenden Versuch soll Kristallviolett (Bestandteil von violetter Tinte) durch Zugabe von Base entfärbt werden. Der Reaktionsverlauf wird photometrisch verfolgt.
Kristallviolett
Das Prisma in einem Photometer zerlegt das Licht aus einer Lichtquelle in seine einzelnen Spektralanteile und der Monochromator des Photometers filtert eine Wellenlänge aus dem Spektrum heraus mit der die Probelösung bestrahlt wird. Das Photometer misst die Absorption (das Maß für das Absorptionsvermögen eines Stoffes), indem es die Intensität des hindurchtretenden Lichts einer gefärbten Probelösung I mit der Intensität einer ungefärbten Lösung Io vergleicht:
E = lg (Io/I)
Nach dem Lambert-Beer'schen Gesetz ist die Extinktion einer Lösung proportional zur Schichtdicke der Küvette, der Konzentration der Lösung und dem stoffspezifischen Extinktionskoeffizient. Die Schichtdicke ist mit 1 cm genormt und kann somit in der Gleichung vernachlässigt werden. Für die Referenzlösung gilt ebenfalls das Lambert-Beer'sche Gesetz und durch Gleichsetzen der Gleichungen für beide Lösungen kann die Konzentration der Probelösung berechnet werden:
c = (E/Eo) · co
Die Reaktion der Entfärbung hängt prinzipiell von den Eduktkonzentrationen ab, die Base liegt jedoch im deutlichen Überschuss vor ([OH−] >> [Kristallviolett]), sodass sich die Konzentration an Lauge vernachlässigbar wenig im Laufe der Reaktion verändert.
Verwenden Sie für den Versuch die im Saal bereitstehende Kristallviolettlösung und nicht die Indikatorlösung!
Notieren Sie sich die Konzentration der Kristallviolettlösung.
Bereiten Sie eine Absorptionsmessung vor, indem sie wie im Abschnitt Aufnahme von Absorptionsspektren beschrieben vorgehen und dabei eine 0.1 m KOH-Lösung gefüllte
Küvette als Referenz verwenden. Messen Sie danach ein Absorptionsspektrum von einigen wenigen Tropfen Kristallviolettlösung in 2 mL Wasser. Lesen Sie aus dem gemessenen Spektrum das Absorptionsmaximum
ab und starten Sie ein Trend-Diagramm, füllen die Menüs entsprechend
aus und geben dabei den Bereich von +/− 5 nm um das zuvor bestimmte Absorptionsmaximum an.
Bereiten Sie nun eine Küvette mit 1 mL Kristallviolettlösung vor.
Die vorhandene Kristallviolettlösung muss verdünnt werden, damit die Reaktion durchgeführt werden kann.
Der Absorptionswert des Trenddiagramms sollte zu Beginn zwischen 0.5 und 1.0 betragen. Testen Sie Verdünnungen von 1:10 (das bedeutet 1 Teil Kristallviolett und 9 Teile Wasser), 1:50 oder 1:100.
Notieren Sie die verwendete Verdünnung und geben Sie sie in der Auswertung an!
Stellen Sie die verdünnte Kristallviolettlösung in den Küvettenhalter und geben sie mit der Mikropipette rasch 2 mL 0.1 m KOH in die Küvette und mischen,
indem Sie den Pipettenkolben vorsichtig auf- und abbewegen, die Reaktionsmischung durch.
Klicken Sie auf den oberen Play-Button und setzen Sie einen Haken bei Strobe/Lamp enable.
Durch Klicken auf den unteren Play-Button starten Sie rasch das Trenddiagramm. Nehmen Sie 10 Minuten lang Messdaten auf, bis die Lösung nahezu vollständig entfärbt ist.
Zur Auswertung werden die Messdaten exportiert und in QtiPlot eingelesen. Der Header wird entfernt. Die erste Spalte beinhaltet lediglich die absolute Uhrzeit der Messung und wird gelöscht. Die beiden verbliebenen Spalten beinhalten die Zeit in Sekunden, die nach Start der Messung vergangen sind und den jeweils zugeordneten Absorptionswert. Der Absorptionswert wird nach erfolgreicher Überprüfung der Reaktionsordnung über der Zeit geplottet. Die Daten werden angefittet.
Berechnen Sie die Geschwindigkeitskonstante k' der Reaktion (welche Einheit hat k'?).
Welche Reaktionsordnung liegt vor? Mit welcher Methode können Sie dies herausfinden? [5P]
Welche Reaktionsordnung ist nach dem Reaktionsmechanismus für die Entfärbung von Kristallviolett zu erwarten? Wie ist dies mit dem Ergebnis Ihres Versuches in Einklang zu bringen? [3P]
Führen Sie eine Literaturrecherche durch und suchen Sie nach k'-Werten für die
Entfärbungsreaktion. Geben Sie für jeden gefunden Referenzwert eine Quelle an!
Vergleichen Sie die gefundenen k'-Werte
mit denen Ihres Versuchs.
Wie lassen sich mögliche Abweichungen erklären? [4P]
Wieso muss für ein genaues Versuchsergebnis eine mit KOH-Lösung gefüllte Küvette als Referenzprobe gemessen werden? [1P]
Hier wird die Absorption gemessen. Der Detektor steht im Strahlengang direkt hinter der Probe. Wird die Fluoreszenz eines Farbstoffes
gemessen, wird der Detektor im 90° Winkel aufgestellt. Wie erklärt sich der Unterschied? [2P]
Diese allgemeinen Konzepte sollte Sie hinter den Versuchen erkennen
Der Ablauf einer chemischen Reaktion lässt sich mit Hilfe von mathematischen Gleichungen beschreiben. Die Reaktionsgeschwindigkeit spiegelt dabei die Konzentrationsabnahme der Edukte und die Konzentrationszunahme der Produkte pro Zeiteinheit wider. Die Reaktionsordnung beschreibt den Exponenten in der Gleichung.